
Analysis of Primary Liquid Chromatography Mass Spectrometry Data by Neural Networks for Plant Samples Classification
 , ,
, ,
Abstract
:1. Introduction
2. Experimental
2.1. Materials and Reagents
2.2. Sample Preparation
2.3. Instrumentation
2.4. Software and Packages
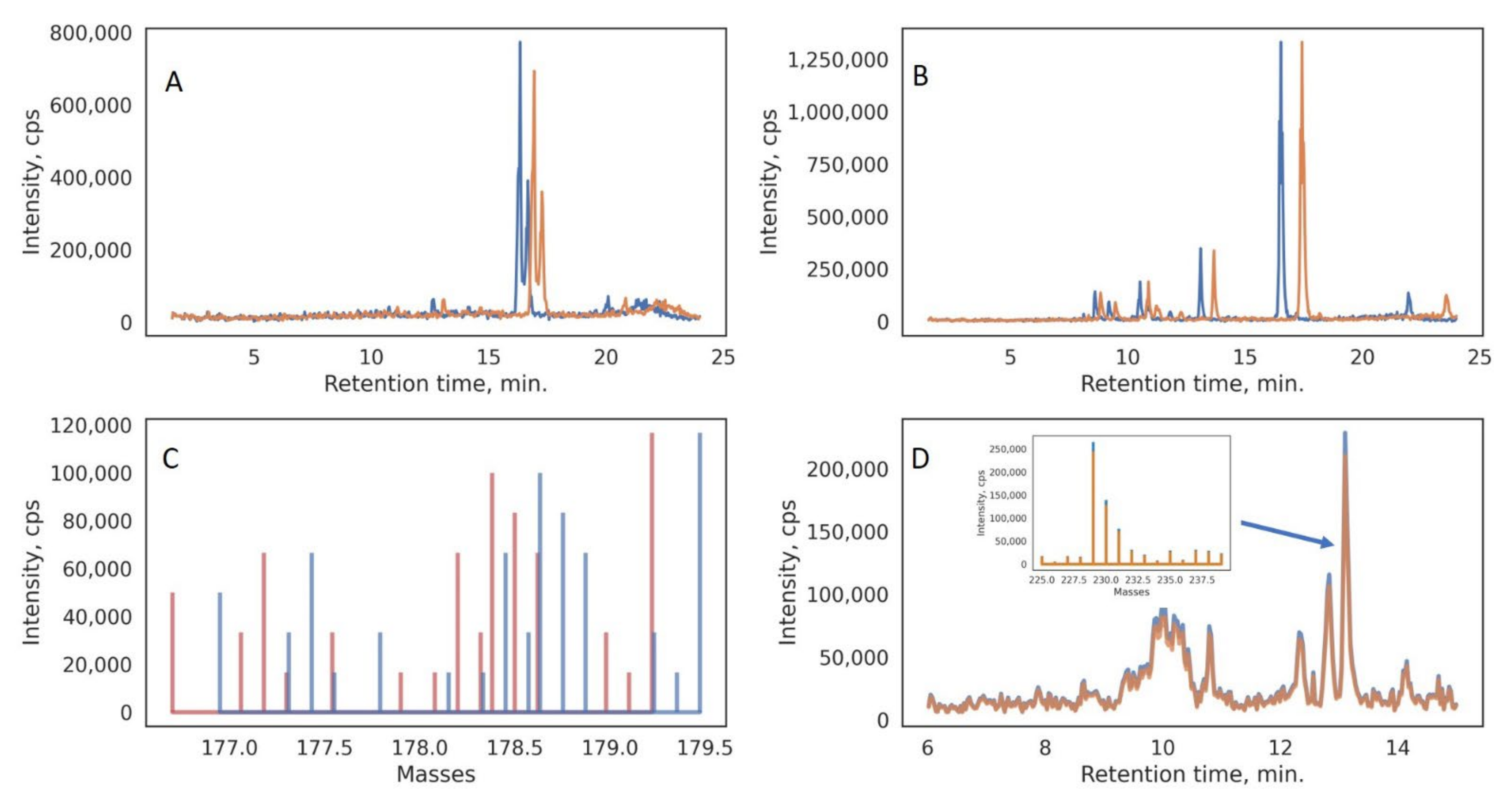
2.5. LC-LRMS Data Treatment
3. Results and Discussion
3.1. LC-MS Data Pretreatment and Augmentation
3.2. Data Analysis Using the SVM
3.3. Class-Important Features from the SVM Model Results

{kind=link}
{kind=link}
{kind=link}
| N° | Retention Time (m/z) | [M + H]+, m/z (Δ, ppm) * | Adduct Ions, m/z | Main MS/MS Fragments, m/z | Annotation | Reference |
|---|---|---|---|---|---|---|
| 1 | 9.0 (305) | 599.1967 (C27H35O15, 0.6) | 621.1785 [M + Na]+ 637.1546 [M + K]+ | 599−467 = Api(f) 467−305 = Glc 305−269 = 2H2O 305−203 = C5H10O2 203−175 = CO 203−159 = CO2 203−147 = 2CO 159−131 = CO | Coumarin-related signal: Api(f)-Glc-heraclenol (or its isomer) | [39] |
| 2 | 6.4 (495) | 457.1338 (C20H25O12, −1.7) | 479.1125 [M + Na]+ 495.0900 [M + K]+ | 457−325 = Api(f) 325−163 = Glc 163−119 = CO2 163−107 = 2CO | Coumarin-related signal: Api(f)-Glc-hydroxycoumarin (Apiosylskimmin) | [40] |
| 3 | 10.4 (607) | 607.2184 C36H31N4O4Mg (−2.0) | 1186.5182 [2M + NH4]+ 1191.4738 [2M + Na]+ 1207.4478 [2M + K]+ | 410.1335 (C21H20N3O6, 2.8) | Chlorophyll-related signal: Tissue-specific protochlorophyllide analog | [42] |
| 4 | 8.9 (603) | 603.2078 (C33H31N4O6Mg, −2.6) | 1178.4983 [2M + NH4]+ 1183.4536 [2M + Na]+ 1199.4277 [2M + K]+ | 440.1416 (C29H18N3O2, −0.8) | Chlorophyll-related signal: Tissue-specific protochlorophyllide analog (e.g., Mg-oxo-purpurin-18) | [42,45] |
| 5 | 21.8 (625, 627) | 625.2662 (C35H37N4O7, −0.8) | 647.2492 [M + Na]+ 663.2235 [M + K]+ | 625−607 = H2O 621−565 = C2H4O2 565−537 = CO | Chlorophyll-related signal: (151-hydroxy-lactone-pheophorbide a or its isomer) | [46] |
| 6 | 22.3 621 (623) | 621.2738 (C36H37N4O6, 4.8) | 643.2533 [M + Na]+ 659.2278 [M + K]+ | 621−593 = CO 621−561 = C2H4O2 561−533 = CO | Chlorophyll-related signal: (Methylpheophorbide b or its isomer) | [46] |
| 7 | 18.7 (522) | 522.3570 (C26H53NO7P, 3.1) | 544.3373 [M + Na]+ | 522−504 = H2O 522−339 = C5H13NO4P 522−184 = C21H38O3 184−166 = H2O 184−124 = C3H10N 184−104 = PO3 104−86 = H2O | Lipid-related signal: PC (18:1) | [47] |
| 8 | 17.0 (520, 521) | 520.3408 (C26H51NO7P, 1.0) | 542.3239 [M + Na]+ | 520−502 = H2O 520−337 = C5H13NO4P 520−184 = C21H36O3 184−166 = H2O 184−124 = C3H10N 184−104 = PO3 104−86 = H2O | Lipid-related signal: PC (18:2) | [47] |
3.4. Specificity of Selected Markers
3.5. Data Analysis Using the Neural Networks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gong, F.; Liang, Y.-Z.; Xie, P.-S.; Chau, F.-T. Information theory applied to chromatographic fingerprint of herbal medicine for quality control. J. Chromatogr. A 2003, 1002, 25–40. [Google Scholar] [CrossRef]
- Bauer, R.; Titiel, G. Quality assessment of herbal preparations as a precondition of pharmacological and clinical studies. Phytomedicine 1996, 2, 193–198. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Z.; Li, Y.; Qian, Z. Overview of the quality standard research of traditional Chinese medicine. Front. Med. 2011, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Alaerts, G.; Dejaegher, B.; Smeyers-Verbeke, J.; Vander Heyden, Y. Recent developments in chromatographic fingerprints from herbal products: Set-up and data analysis. Comb. Chem. High Throughput Screen 2010, 13, 900–922. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.N.; Liu, Y.; Kokot, S. Two-dimensional fingerprinting approach for comparison of complex substances analysed by HPLC-UV and fluorescence detection. Analyst 2011, 136, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Megson, D.; Brown, T.A.; Johnson, G.W.; O’Sullivan, G.; Bicknell, A.W.J.; Votier, S.C.; Lohan, M.C.; Comber, S.; Kalin, P.; Worsfold, P.J. Identifying the provenance of Leach’s storm petrels in the North Atlantic using polychlorinated biphenyl signatures derived from comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry. Chemosphere 2014, 114, 195–202. [Google Scholar] [CrossRef]
- Zhou, W.; Xie, M.-F.; Zhang, X.-Y.; Liu, T.-T.; Yu, Y.-J.; Duan, G.-L. Improved liquid chromatography fingerprint of fat-soluble Radix isatidis extract using multi-wavelength combination technique. J. Sep. Sci. 2011, 34, 1123–1132. [Google Scholar] [CrossRef]
- Wang, Y.; Li, B.H.; Ni, Y.N.; Kokot, S. Multi-wavelength high-performance liquid chromatography: An improved method for analysis of complex substances such as Radix Paeoniae herbs. Chemom. Intell. Lab. Syst. 2014, 130, 159–165. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Dai, R.; Yu, Y.; Saeed, M.K.; Li, L.; Meng, W.; Zhang, X. Chromatographic fingerprint analysis of Cephalotaxus sinensis from various sources by high-performance liquid chromatography–diodearray detection–electrospray ionization-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2007, 45, 38–46. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Luo, Q.; Lu, H.; Liang, Y. Evaluation and prediction of the antioxidant activity of Epimedium from multi-wavelength chromatographic fingerprints and chemometrics. Anal. Methods 2014, 6, 1036–1043. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, G. Assessment of quality consistency in traditional Chinese medicine using multi-wavelength fusion profiling by integrated quantitative fingerprint method: Niuhuang Jiedu pill as an example. J. Sep. Sci. 2019, 42, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, G.-X.; Jin, Y.; Xie, X.-M.; Liu, Y.-C.; Ma, D.-D.; Zhang, J.; Gao, J.-Y.; Li, Y.-F. Holistic evaluation of San-Huang Tablets using a combination of multi-wavelength quantitative fingerprinting and radical-scavenging assays. Chin. J. Nat. Med. 2017, 15, 310–320. [Google Scholar] [CrossRef]
- Li, B.Q.; Chen, J.; Li, J.J.; Wang, X.; Zhai, H.L. The application of a Tchebichef moment method to the quantitative analysis of multiple compounds based on three-dimensional HPLC fingerprint spectra. Analyst 2015, 140, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, B.Q.; Xu, M.L.; Liu, J.J.; Zhai, H.L. Quality assessment of Traditional Chinese Medicine using HPLC-PAD combined with Tchebichef image moments. J. Chromatogr. B 2017, 1040, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Xie, J.; Tang, H.; Shi, C.; Xie, Y.; He, J.; Zeng, Y.; Zhou, H.; Xia, B.; Zhang, C.; et al. Fingerprints of volatile flavor compounds from southern stinky tofu brine with headspace solid-phase microextraction/gas chromatography–mass spectrometry and chemometric methods. Food Sci. Nutr. 2019, 7, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.N.; Mei, M.G.; Kokot, S. One- and two-dimensional gas chromatography–mass spectrometry and high performance liquid chromatography–diode-array detector fingerprints of complex substances: A comparison of classification performance of similar, complex Rhizoma Curcumae samples with the aid of chemometrics. Anal. Chim. Acta 2012, 712, 37–44. [Google Scholar] [CrossRef]
- Song, J.J.; Fang, G.Z.; Zhang, Y.; Deng, Q.; Wang, S. Fingerprint analysis of Ginkgo biloba leaves and related health foods by high-performance liquid chromatography/electrospray ionization-mass spectrometry. J. AOAC Int. 2010, 93, 1798–1805. [Google Scholar] [CrossRef] [Green Version]
- Guijarro-Díez, M.; Nozal, L.; Marina, M.L.; Crego, A.L. Metabolomic fingerprinting of saffron by LC/MS: Novel authenticity markers. Anal. Bioanal. Chem. 2015, 407, 7197–7213. [Google Scholar] [CrossRef]
- Wolfender, J.-L.; Rudaz, S.; Choi, Y.H.; Kim, H.K. Plant metabolomics: From holistic data to relevant biomarkers. Curr. Med. Chem. 2013, 20, 1056–1090. [Google Scholar] [CrossRef]
- Skarysz, A.; Salman, D.; Eddleston, M.; Sykora, M.; Hunsicker, E.; Nailon, W.H.; Darnley, K.; McLaren, D.B.; Thomas, C.L.P.; Soltoggio, A. Fast and automated biomarker detection in breath samples with machine learning. PLoS ONE 2022, 17, e0265399. [Google Scholar] [CrossRef]
- Petrovsky, D.V.; Kopylov, A.T.; Rudnev, V.R.; Stepanov, A.A.; Kulikova, L.I.; Malsagova, K.A.; Kaysheva, A.L. Managing of unassigned mass spectrometric data by neural network for cancer phenotypes classification. J. Pers. Med. 2021, 11, 1288. [Google Scholar] [CrossRef] [PubMed]
- Döll, S.; Farahani-Kofoet, R.D.; Zrenner, R.; Henze, A.; Witzel, K. Tissue-specific signatures of metabolites and proteins in asparagus roots and exudates. Hortic. Res. 2021, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Dossou, S.S.K.; Xu, F.; Cui, X.; Sheng, C.; Zhou, R.; You, J.; Tozo, K.; Wang, L. Comparative metabolomics analysis of different sesame (Sesamum indicum L.) tissues reveals a tissue-specific accumulation of metabolites. BMC Plant Biol. 2021, 21, 352. [Google Scholar] [CrossRef]
- Imai, A.; Lankin, D.C.; Gödecke, T.; Chen, S.-N.; Pauli, G.F. Differentiation of Actaea species by NMR metabolomics analysis. Fitoterapia 2020, 146, 104686. [Google Scholar] [CrossRef]
- In, G.; Seo, H.K.; Park, H.W.; Jang, K.H. A metabolomic approach for the discrimination of red ginseng root parts and targeted validation. Molecules 2017, 22, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turova, P.; Styles, I.; Timashev, V.; Kravets, K.; Grechnikov, A.; Lyskov, D.; Samigullin, T.; Podolskiy, I.; Shpigun, O.; Stavrianidi, A. Unsupervised methods in LC-MS data treatment: Application for potential chemotaxonomic markers search. J. Pharm. Biomed. Anal. 2021, 206, 114382. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, J.; Etmann, C.; Boskamp, T.; Casadonte, R.; Kriegsmann, J.; Maaß, P. Deep learning for tumor classification in imaging mass spectrometry. Bioinformatics 2018, 34, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Bald, T.; Barth, J.; Niehues, A.; Specht, M.; Hippler, M.; Fufezan, C. PymzML-Python module for high throughput bioinformatics on mass spectrometry data. Bioinformatics 2012, 28, 1052–1053. [Google Scholar] [CrossRef] [Green Version]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32, 8024–8035. [Google Scholar]
- Pedregosa, F.; Michel, V.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Vanderplas, J.; Cournapeau, D.; Pedregosa, F.; Varoquaux, G. Scikit-Learn: Machine Learning in Python. J. Machine Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Hunter, J. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Bradski, G.; Kaehler, A. The OpenCV Library. Dr. Dobb’s J. Softw. Tools 2000, 3, 120. [Google Scholar]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A next-generation hyperparameter optimization framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar] [CrossRef]
- Fraga, C.G.; Prazen, B.J.; Synovec, R.E. Comprehensive two-dimensional gas chromatography and chemometrics for the high-speed quantitative analysis of aromatic isomers in a jet fuel using the standard addition method and an objective retention time alignment algorithm. Anal. Chem. 2000, 72, 4154–4162. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Numonov, S.; Bobakulov, K.; Numonova, M.; Sharopov, F.; Setzer, W.N.; Khalilov, Q.; Begmatov, N.; Habasi, M.; Aisa, H.A. New coumarin from the roots of Prangos pabularia. Nat. Prod. Res. 2018, 32, 2325–2332. [Google Scholar] [CrossRef]
- Liu, G.-Q.; Dong, J.; Wang, H.; Hashi, Y.; Chen, S.-Z. Differentiation of four pairs of furocoumarin isomers by electrospray ionization tandem mass spectrometry. Eur. J. Mass Spectrom. 2010, 16, 215–220. [Google Scholar] [CrossRef]
- Niu, X.-M.; Li, S.-H.; Wu, L.-X.; Li, L.; Gao, L.-H.; Sun, H.-D. Two new coumarin derivatives from the roots of Heracleum rapula. Planta Med. 2004, 70, 578–581. [Google Scholar] [CrossRef]
- Luo, L.; Liu, X.; Jin, X.; Liu, Y.; Ma, J.; Zhang, S.; Zhang, D.; Chen, X.; Sheng, L.; Li, Y. Simultaneous determination of skimmin, apiosylskimmin, 7-hydroxycoumarin and 7-hydroxycoumarin glucuronide in rat plasma by liquid chromatography–orbitrap mass spectrometry and its application to pharmacokinetics. Biomed. Chromatogr. 2022, 36, e5223. [Google Scholar] [CrossRef]
- Curini, M.; Cravotto, G.; Epifano, F.; Giannone, G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. [Google Scholar] [CrossRef]
- Müller, T.; Vergeiner, S.; Kräutler, B. Structure elucidation of chlorophyll catabolites (phyllobilins) by ESI-mass spectrometry—Pseudo-molecular ions and fragmentation analysis of a nonfluorescent chlorophyll catabolite (NCC). Int. J. Mass Spectrom. 2014, 365–366, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Skribanek, A.; Apatini, D.; Inaoka, M.; Boddi, B. Protochlorophyllide and chlorophyll forms in dark-grown stems and stem-related organs. J. Photochem. Photobiol. B Biol. 2000, 55, 172–177. [Google Scholar] [CrossRef]
- Skribanek, A.; Boka, K.; Boddi, B. Tissue specific protochlorophyll(ide) forms in dark-forced shoots of grapevine (Vitis vinifera L.). Photosynth. Res. 2004, 82, 141–150. [Google Scholar] [CrossRef]
- Drogat, D.; Barrière, M.; Granet, R.; Sol, V.; Krausz, P. High yield preparation of purpurin-18 from Spirulina maxima. Dyes Pigm. 2011, 88, 125–127. [Google Scholar] [CrossRef]
- Viera, I.; Roca, M.; Perez-Galvez, A. Mass spectrometry of non-allomerized chlorophylls a and b derivatives from plants. Curr. Org. Chem. 2018, 22, 842–876. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; Milic, I.; Fedorova, M. Identification of carbonylated lipids from different phospholipid classes by shotgun and LC-MS lipidomics. Anal. Bioanal. Chem. 2015, 407, 5161–5173. [Google Scholar] [CrossRef]



| Species | Plant Parts (#) | Specimen’s Voucher |
|---|---|---|
| Prangos pabularia | Leaves (1.1), Fruits (1.2), Stems (1.3) | MW0858238 |
| Ferulago phialocarpa | Stems (2.1), Leaves (2.2) | 031-IR-19 |
| Cachrys libanotis | Leaves (3.1), Inflorescence (3.2), Roots (3.3) | MW0798144 |
| Prangos acaulis | Leaves (4.1), Roots (4.2), Fruits (4.3) | MW0744005 |
| Prangos ferulacea | Stems (5.1), Fruits (5.2) | MW0751912 |
| Prangos didyma | Fruits (6.1), Stems (6.2) | MW0857912 |
| Ferulago subvelutina | Stems (7.1), Leaves (7.2), Inflorescence (7.3) | 098-IR-19 |
| Prangos ammophila | Leaves (8.1), Roots (8.2), Inflorescence (8.3) | MW0857867 |
| Prangos trifida | Leaves (9.1), Fruits (9.2), Stems (9.3) | MW0798580 |
| Ferulago angulata | Stems (10.1), Leaves (10.2), Roots (10.3) | 085-IR-19 |
| Cachrys sicula | Inflorescence (11.1), Leaves (11.2), Stems (11.3), Roots (11.4) | MW0798143 |
| Prangos chelantofolia | Fruits (12.1), Roots (12.2) | MW0753034 |
| Ferulago contracta | Leaves (13.1), Stems (13.2) | 053-IR-19 |
| Cachrys pungens | Fruits (14.1), Leaves (14.2) | MW0784701 |
| Bilacunaria microcapra | Roots (15.1), Leaves (15.2), Fruits (15.3), Stems (15.4) | 018-IR-19 |
| Diplotaenia cachrydifolia | Inflorescence (16.1), Leaves (16.2), Stems (16.3), Roots (16.4) | 164-IR-19 |
| Bilacunaria microcapra | Leaves (17.1), Roots (17.2), Stems (17.3), Fruits (17.4) | 162-IR-19 |
| Ferulago phialocarpa | Roots (18.1), Leaves (18.2) | 169-IR-19 |
| Azilia eryngioides | Roots (19.1), Leaves (19.2), Stems (19.3) | 167-IR-19 |
| Seseli olivieri | Stems (20.1), Leaves (20.2) | 173-IR-19 |
| Prangos crossoptera | Fruits (21.1), Leaves (21.2) | MW0753036 |
| Bilacunaria microcapra | Leaves (22.1), Inflorescence (22.2) | 028-IR-19 |
| Seseli ghafoorianum | Stems (23.1), Leaves (23.2) | 124-IR-19 |
| Type of Augmentation | Description | Variation Range (Step Size) | Real Process during Experiment | Augmentation Coefficient |
|---|---|---|---|---|
| Chromatogram stretching along the entire retention time axis (A) | Each mass-chromatogram was stretched by adding of new time points with intermediate signal intensity values | ±30 (10) timepoints * | Wrong pump calibration (incorrect flow rate) | Nsamp × 6 |
| Gradient chromatogram stretching along the retention time axis (B) | Each mass-chromatogram was split into 15 segments (50 timepoints) and similar stretching procedure was applied to each segment with increasing number of inserted timepoints | From 1 to 7 points for each segment | Wrong gradient program or insufficient flow from organic phase pump | Nsamp |
| Gradient chromatogram shrinkage along the retention time axis (C) | Each mass-chromatogram was split into 15 segments (50 timepoints) and shrinkage procedure was applied to each segment with increasing number of inserted timepoints | From 1 to 7 points for each segment | Wrong gradient program or insufficient flow from water phase pump | Nsamp |
| Mass shifts (D) | Each raw m/z value in each spectrum is shifted to a specific Δ. This Δ is smaller for low masses and bigger for high masses (linear dependence). | (1) From ±0.1 Da to ±0.6 Da(2) From ±0.2 Da to ±1 Da | Wrong quadrupole calibration | Nsamp × 4 |
| Intensity alteration (E) | All signal intensities are either reduced or enhanced by a specified value | ±5% (5%) | Problems with ESI source or detector gain variations | Nsamp × 2 |
| Augmentation Type | Precision (mean ± SD) * | Recall (mean ± SD) | F1-Score (mean ± SD) |
|---|---|---|---|
| No augmentation | 0.72 ± 0.07 | 0.68 ± 0.08 | 0.68 ± 0.08 |
| Chromatogram stretching (A) | 0.75 ± 0.10 | 0.72 ± 0.12 | 0.73 ± 0.12 |
| Gradient stretching (B) | 0.74 ± 0.07 | 0.70 ± 0.10 | 0.70 ± 0.09 |
| Gradient shrinkage (C) | 0.73 ± 0.06 | 0.70 ± 0.09 | 0.69 ± 0.10 |
| Mass shifts (D) | 0.74 ± 0.04 | 0.69 ± 0.06 | 0.70 ± 0.05 |
| Intensity alteration (E) | 0.72 ± 0.01 | 0.68 ± 0.03 | 0.68 ± 0.03 |
| Full augmentation (A–E) | 0.77 ± 0.02 | 0.75 ± 0.03 | 0.74 ± 0.03 |
| Results for the Whole Dataset | |||
|---|---|---|---|
| Metric | SVM | CNN | SNN |
| Precision | 0.77 ± 0.02 | 0.81 ± 0.05 | 0.81 ± 0.03 |
| Recall | 0.75 ± 0.03 | 0.77 ± 0.06 | 0.78 ± 0.04 |
| F1 | 0.74 ± 0.03 | 0.76 ± 0.07 | 0.77 ± 0.05 |
| F1-score for plant parts classes | |||
| Part | SVM | CNN | SNN |
| Roots (33) * | 0.55 | 0.75 | 0.76 |
| Stems (45) | 0.89 | 0.83 | 0.89 |
| Leaves (60) | 0.94 | 0.94 | 0.88 |
| Fruits (48) | 0.71 | 0.77 | 0.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turova, P.; Stavrianidi, A.; Svekolkin, V.; Lyskov, D.; Podolskiy, I.; Rodin, I.; Shpigun, O.; Buryak, A. Analysis of Primary Liquid Chromatography Mass Spectrometry Data by Neural Networks for Plant Samples Classification. Metabolites 2022, 12, 993. https://doi.org/10.3390/metabo12100993
Turova P, Stavrianidi A, Svekolkin V, Lyskov D, Podolskiy I, Rodin I, Shpigun O, Buryak A. Analysis of Primary Liquid Chromatography Mass Spectrometry Data by Neural Networks for Plant Samples Classification. Metabolites. 2022; 12(10):993. https://doi.org/10.3390/metabo12100993
Chicago/Turabian StyleTurova, Polina, Andrey Stavrianidi, Viktor Svekolkin, Dmitry Lyskov, Ilya Podolskiy, Igor Rodin, Oleg Shpigun, and Aleksey Buryak. 2022. "Analysis of Primary Liquid Chromatography Mass Spectrometry Data by Neural Networks for Plant Samples Classification" Metabolites 12, no. 10: 993. https://doi.org/10.3390/metabo12100993