
Virome Analysis of Signal Crayfish (Pacifastacus leniusculus) along Its Invasion Range Reveals Diverse and Divergent RNA Viruses


Abstract
:1. Introduction
2. Materials and Methods
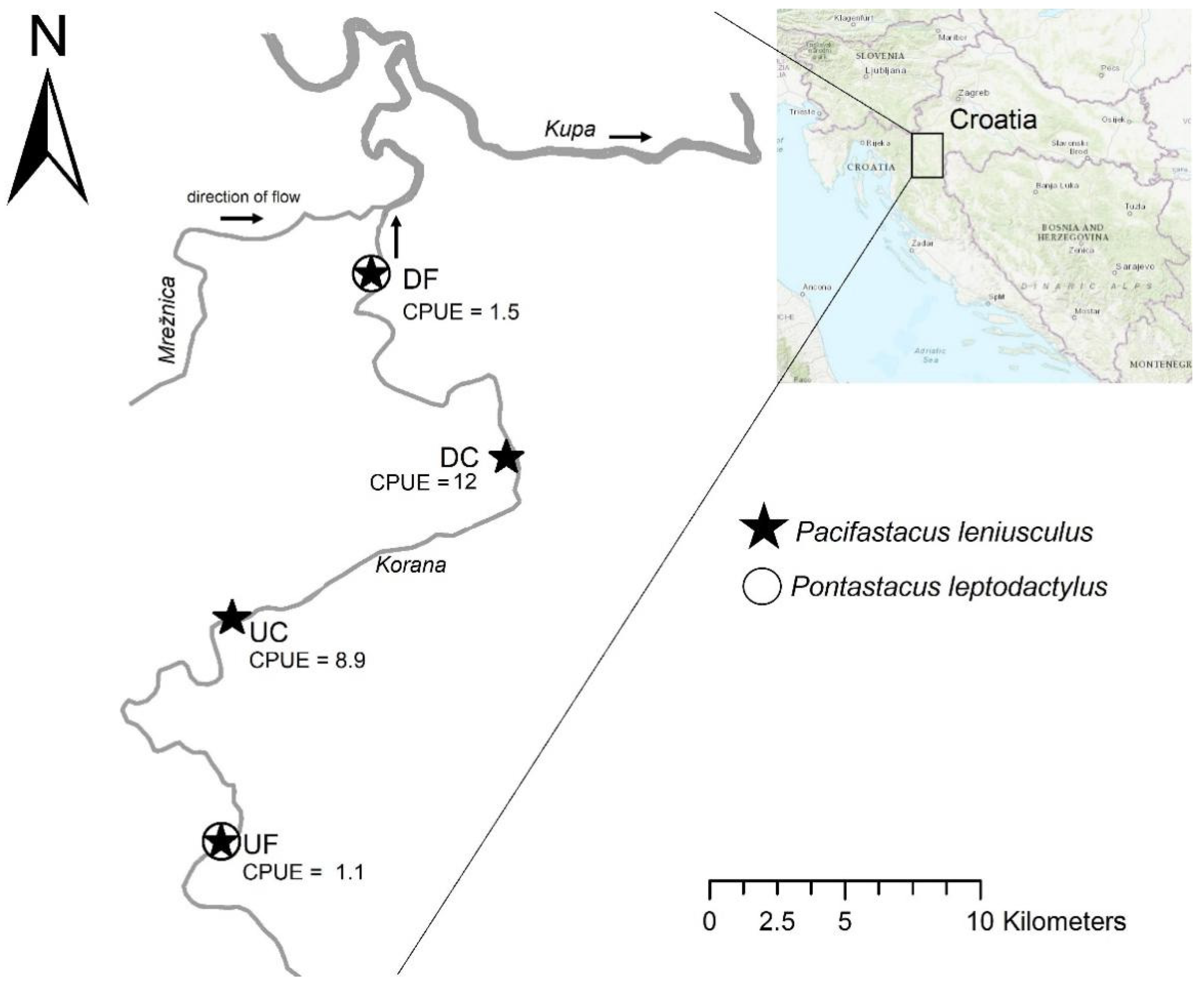
2.1. Study Area and Sample Collection
2.2. RNA Extraction and Sequencing
2.3. Bioinformatic Analysis for Detection of Viral Sequences
2.4. PCR Detection of Selected Viral Contigs in Hepatopancreas Samples
2.5. Phylogenetic Analyses
2.6. Analysis of Nucleotide Diversities
3. Results and Discussion
3.1. Novel Viral Sequences Identified in Hepatopancreas of Signal Crayfish
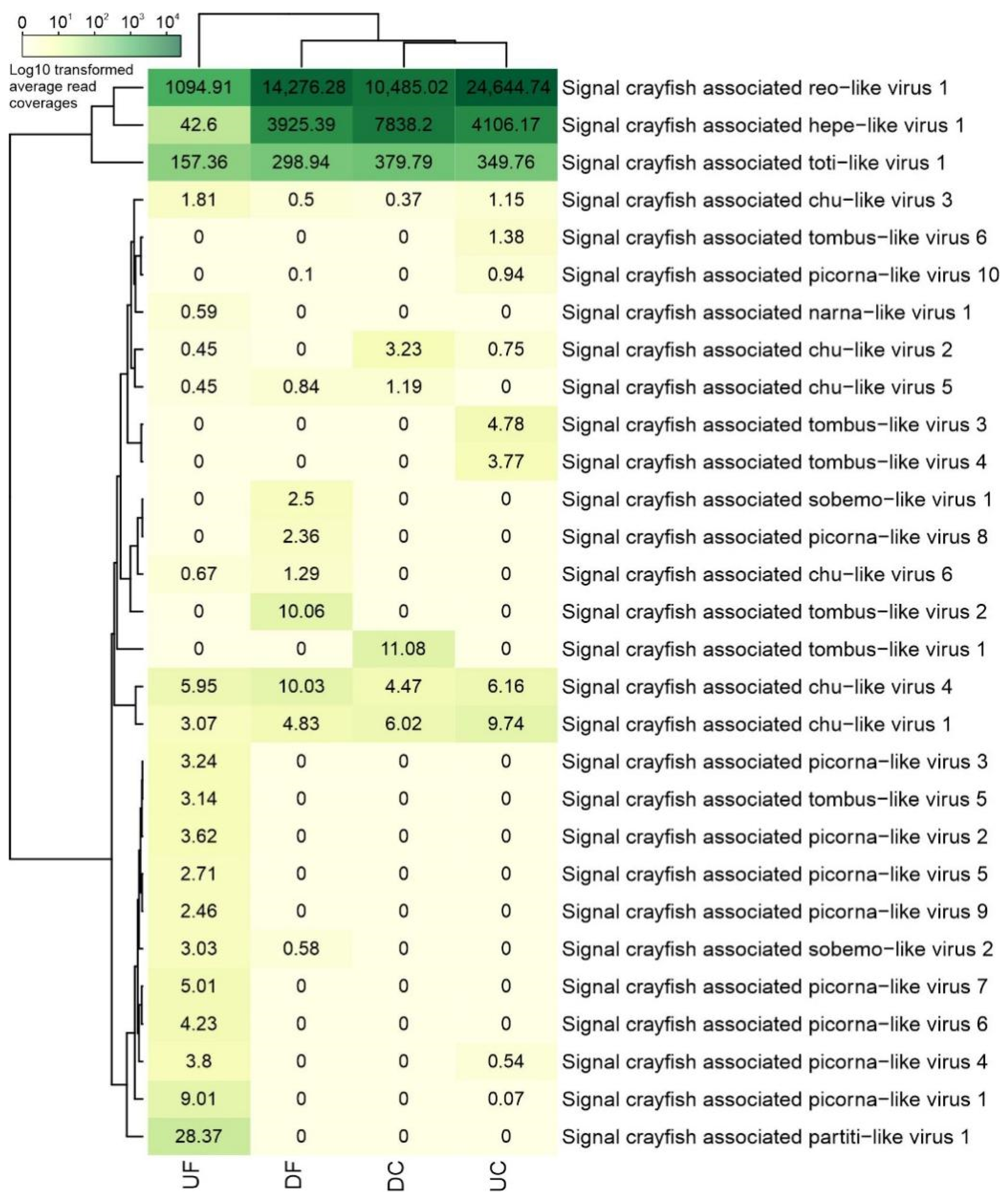
3.1.1. Overview of the Newly Identified Viral Sequences in Hepatopancreas of Signal Crayfish
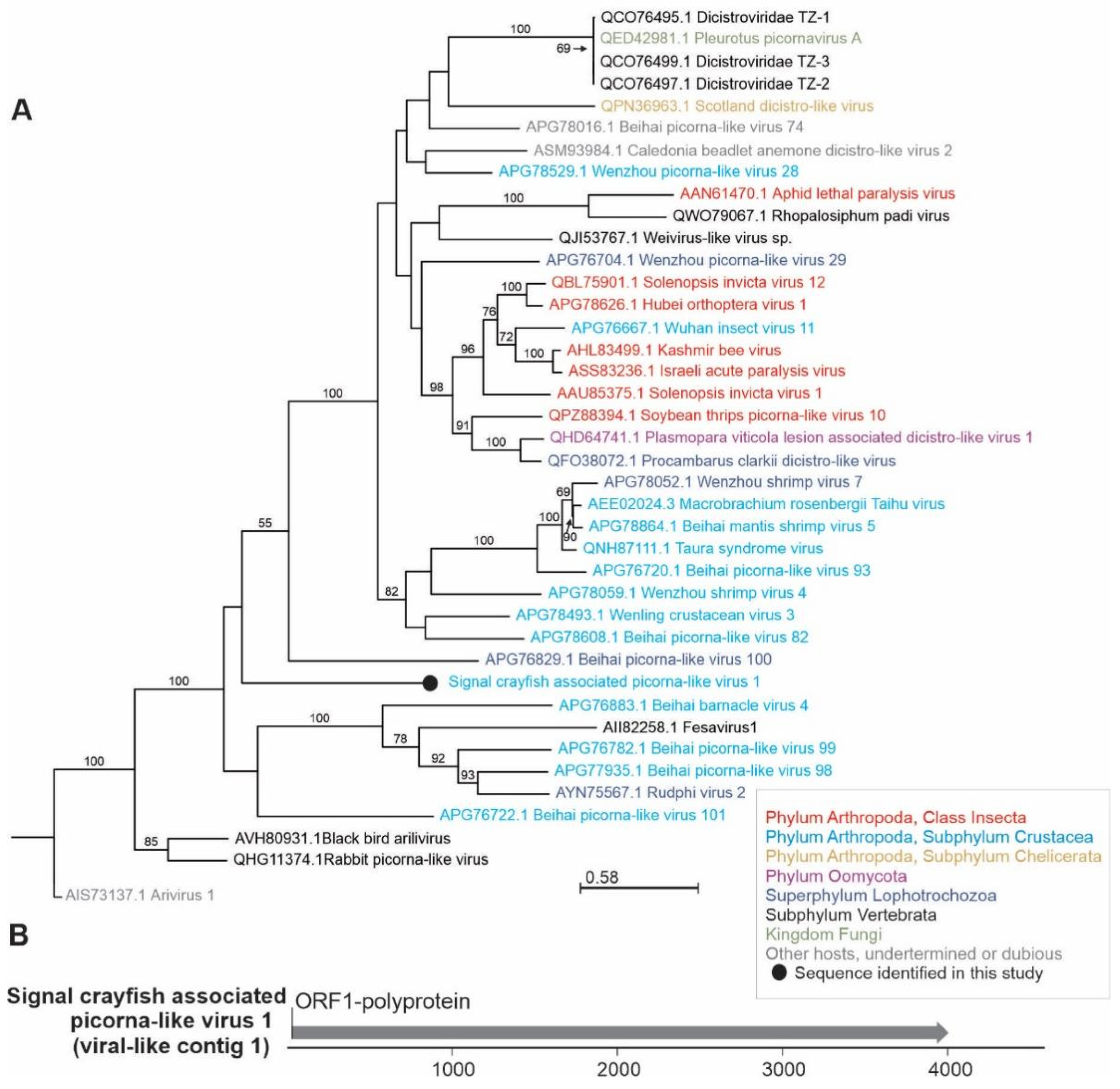
3.1.2. Phylogenetic Relationships and Genome Organization of Selected Newly Identified Viral Sequences
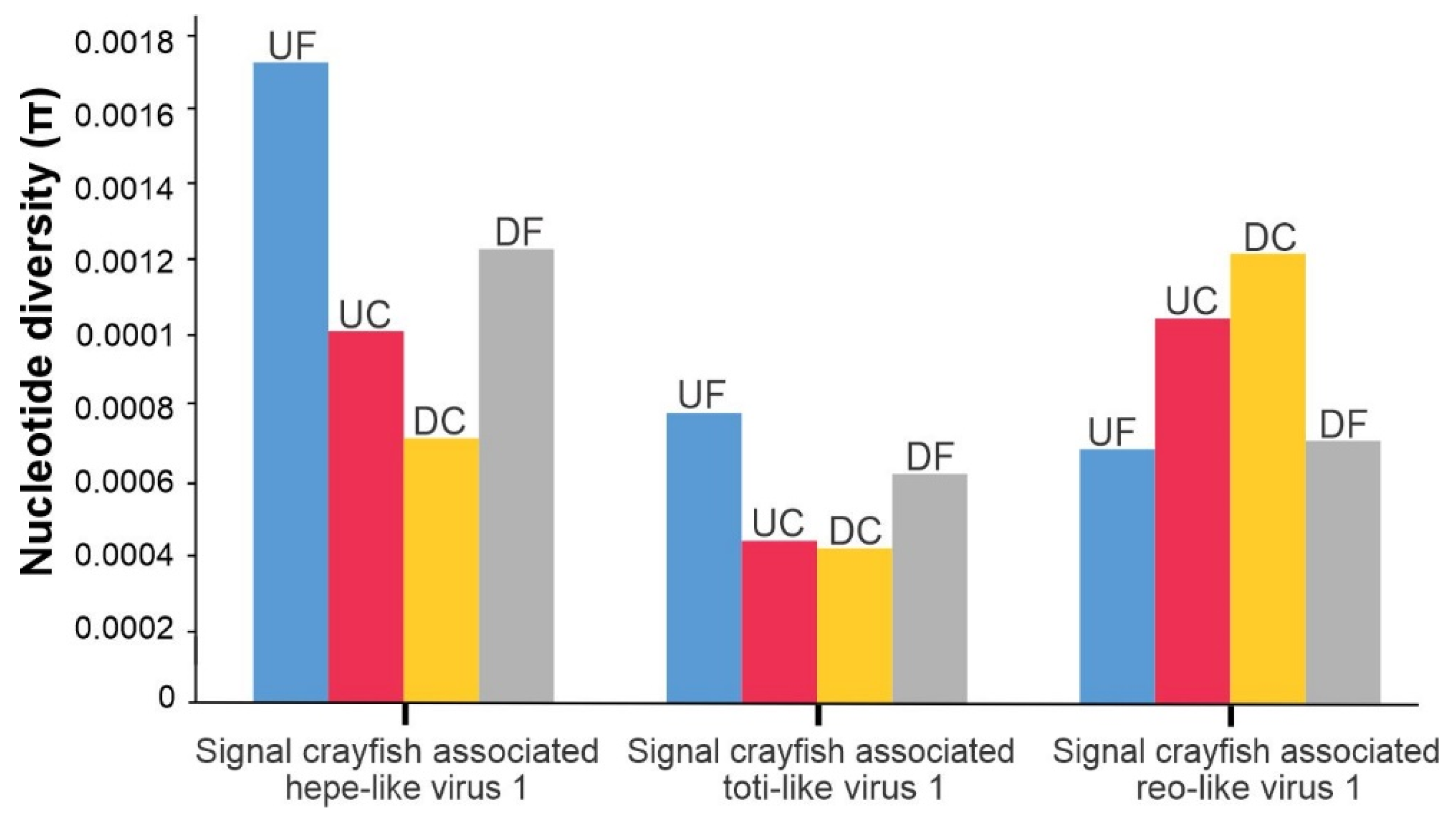
3.2. Abundance of Viral Sequences and Nucleotide Diversity of Selected Virus Populations along the Invasion Range
4. Conclusions
- We reported, for the first time, the virome of signal crayfish hepatopancreas tissue and found a high diversity of novel divergent viral sequences most similar to different unclassified RNA viruses.
- We identified putative novel RNA viruses, including near complete genome sequence of signal crayfish-associated hepe-like virus 1 and toti-like virus 1, and the partial genomes of signal crayfish-associated reo-like virus 1 and picorna-like viruses. We identified additional tombus-like, partiti-like, and chu-like virus sequences potentially representing novel crayfish viruses. This pioneer study represents a baseline for the future research of a signal crayfish virome, e.g., to confirm the association of novel viruses with signal crayfish host, and to investigate their potential involvement in the observed necrotizing hepatopancreatitis.
- We speculate that the differences in the signal crayfish population density along the invasion range, non-random dispersal, and possibilities of inter-specific viral transmissions may have an effect on the diversity and abundance of signal crayfish-associated viral sequences. Different hypotheses can be postulated to explain these patterns, and this study represents a baseline for the further research of virus transmission dynamics as a result of the invader’s fast dispersal, including inter-species transmission between the signal crayfish as an invader and P. leptodactylus as a co-occurring and phylogenetically related native species.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 2015, 1–26. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J.; et al. Abundant and Diverse RNA Viruses in Insects Revealed by RNA-Seq Analysis: Ecological and Evolutionary Implications. Msystems 2020, 5, e00039-20. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Hirai, J.; Hunt, B.P.V.; Suttle, C.A. Arthropods and the evolution of RNA viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef]
- Zhang, Z.-Q. Phylum Arthropoda. In Animal Biodiversity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness; Zootaxa: Auckland, New Zealand, 2013; Volume 3703, p. 17. [Google Scholar]
- Reynolds, J.; Souty-Grosset, C.; Richardson, A. Ecological roles of crayfish in freshwater and terrestrial habitats. Freshw. Crayfish 2013, 19, 197–218. [Google Scholar] [CrossRef]
- Holdich, D.M.; Reynolds, J.D.; Souty-Grosset, C.; Sibley, P.J. A review of the ever increasing threat to European crayfish from non-indigenous crayfish species. Knowl. Manag. Aquat. Ecosyst. 2009, 2009, 394–395. [Google Scholar] [CrossRef] [Green Version]
- Lodge, D.M.; Deines, A.; Gherardi, F.; Yeo, D.C.J.; Arcella, T.; Baldridge, A.K.; Barnes, M.A.; Lindsay Chadderton, W.; Feder, J.L.; Gantz, C.A.; et al. Global introductions of crayfishes: Evaluating the impact of species invasions on ecosystem services. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 449–472. [Google Scholar] [CrossRef]
- Twardochleb, L.A.; Olden, J.D.; Larson, E.R. A global meta-analysis of the ecological impacts of nonnative crayfish. Freshw. Sci. 2013, 32, 1367–1382. [Google Scholar] [CrossRef]
- Souty-Grosset, C.; Grandjean, F.; Gouin, N. Conservation and management of native crayfish populations. Freshw. Crayfish 2004, 14, 1–20. [Google Scholar]
- Richman, N.I.; Böhm, M.; Adams, S.B.; Alvarez, F.; Bergey, E.A.; Bunn, J.J.S.; Burnham, Q.; Cordeiro, J.; Coughran, J.; Crandall, K.A.; et al. Multiple drivers of decline in the global status of freshwater crayfish (Decapoda: Astacidea). Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Crowl, T.A.; Crist, T.O.; Parmenter, R.R.; Belovsky, G.; Lugo, A.E. The spread of invasive species and infectious disease as drivers of ecosystem change. Front. Ecol. Environ. 2008, 6, 238–246. [Google Scholar] [CrossRef]
- Van der Veer, G.; Nentwig, W. Environmental and economic impact assessment of alien and invasive fish species in Europe using the generic impact scoring system. Ecol. Freshw. Fish 2015, 24, 646–656. [Google Scholar] [CrossRef]
- Unestam, T.; Weiss, D.W. The host-parasite relationship between freshwater crayfish and the crayfish disease fungus Aphanomyces astaci: Responses to infection by a susceptible and a resistant species. J. Gen. Microbiol. 1970, 60, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Torrijos, L.; Martínez-Ríos, M.; Casabella-Herrero, G.; Adams, S.B.; Jackson, C.R.; Diéguez-Uribeondo, J. Tracing the origin of the crayfish plague pathogen, Aphanomyces astaci, to the Southeastern United States. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Gui, J.F. Virus genomes and virus-host interactions in aquaculture animals. Sci. China Life Sci. 2015, 58, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Geoghegan, J.L.; Di Giallonardo, F.; Cousins, K.; Shi, M.; Williamson, J.E.; Holmes, E.C. Hidden diversity and evolution of viruses in market fish. Virus Evol. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Dragičević, P.; Bielen, A.; Petrić, I.; Hudina, S. Microbial pathogens of freshwater crayfish: A critical review and systematization of the existing data with directions for future research. J. Fish Dis. 2020, 44, 221–247. [Google Scholar] [CrossRef]
- Sánchez-Paz, A. White spot syndrome virus: An overview on an emergent concern. Vet. Res. 2010, 41, 43. [Google Scholar] [CrossRef] [Green Version]
- Longshaw, M. Parasites, Commensals, Pathogens and Diseases of Crayfish. In Biology and Ecology of Crayfish; Stebbing, M., Longshaw, P., Eds.; Taylor & Francis Group, LLC: Boca Raton, FL, USA, 2016; pp. 171–250. [Google Scholar]
- Bateman, K.S.; Stentiford, G.D. A taxonomic review of viruses infecting crustaceans with an emphasis on wild hosts. J. Invertebr. Pathol. 2017, 147, 86–110. [Google Scholar] [CrossRef]
- Grandjean, F.; Gilbert, C.; Razafimafondy, F.; Vucić, M.; Delaunay, C.; Gindre, P.; Bouchard, J.; Raimond, M.; Moumen, B. A new bunya-like virus associated with mass mortality of white-clawed crayfish in the wild. Virology 2019, 533, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouba, A.; Petrusek, A.; Kozák, P. Continental-wide distribution of crayfish species in Europe: Update and maps. Knowl. Manag. Aquat. Ecosyst. 2014, 5, 413. [Google Scholar] [CrossRef]
- Hudina, S.; Faller, M.; Lucić, A.; Klobučar, G.; Maguire, I. Distribution and dispersal of two invasive crayfish species in the Drava River basin, Croatia. Knowl. Manag. Aquat. Ecosyst. 2009, 9, 394–395. [Google Scholar] [CrossRef] [Green Version]
- Dragičević, P.; Faller, M.; Kutleša, P.; Hudina, S. Update on the signal crayfish, pacifastacus leniusculus (Dana, 1852) range expansion in Croatia: A 10-year report. BioInvasions Rec. 2020, 9, 793–807. [Google Scholar] [CrossRef]
- Hudina, S.; Žganec, K.; Lucić, A.; Trgovčić, K.; Maguire, I. Recent invasion of the karstic river systems in croatia through illegal introductions of the signal crayfish. Freshw. Crayfish 2013, 19, 21–27. [Google Scholar] [CrossRef]
- Hudina, S.; Kutleša, P.; Trgovčić, K.; Duplić, A. Dynamics of range expansion of the signal crayfish (Pacifastacus leniusculus) in a recently invaded region in Croatia. Aquat. Invasions 2017, 12, 67–75. [Google Scholar] [CrossRef]
- Anderson, R.M.; May, R.M. Coevolution of Hosts and Parasites. Parasitology 1982, 85, 411–426. [Google Scholar] [CrossRef]
- Costa, V.A.; Mifsud, J.C.O.; Gilligan, D.; Williamson, J.E.; Holmes, E.C.; Geoghegan, J.L. Metagenomic sequencing reveals a lack of virus exchange between native and invasive freshwater fish across the Murray-Darling Basin, Australia. Virus Evol. 2021, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Westman, K. A new folding trap model which prevents crayfish from escaping. Freshw. Crayfish 1978, 4, 235–242. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Gorska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2021. Available online: https://www.R-project.org (accessed on 9 November 2021).
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, R.; Serra, F.; Tárraga, J.; Medina, I.; Carbonell, J.; Pulido, L.; De María, A.; Capella-Gutíerrez, S.; Huerta-Cepas, J.; Gabaldón, T.; et al. Phylemon 2.0: A suite of web-tools for molecular evolution, phylogenetics, phylogenomics and hypotheses testing. Nucleic Acids Res. 2011, 39, 470–474. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Nelson, C.W.; Moncla, L.H.; Hughes, A.L. SNPGenie: Estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics 2015, 31, 3709–3711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, X.; Taylor, A.; Jiao, C.; Xu, Y.; Cai, X.; Wang, X.; Ge, C.; Pan, G.; Wang, Q.; et al. Diversity, Distribution, and Evolution of Tomato Viruses in China Uncovered by Small RNA Sequencing. J. Virol. 2017, 91, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakijkosol, O.; Jaroenram, W.; Owens, L.; Elliman, J. Reverse transcription polymerase chain reaction (RT-PCR) detection for Australian Cherax reovirus from redclaw crayfish (Cherax quadricarinatus). Aquaculture 2021, 530, 735881. [Google Scholar] [CrossRef]
- Hayakijkosol, O.; Owens, L. Investigation into the pathogenicity of reovirus to juvenile Cherax quadricarinatus. Aquaculture 2011, 316, 1–5. [Google Scholar] [CrossRef]
- Bekavac, A.; Dragičević, P.; Dragun, Z.; Beck, A.; Maguire, I.; Ivanković, D.; Fiket, Ž.; Gračan, R.; Hudina, S. Disturbance in invasion? Idiopathic necrotizing hepatopancreatitis in the signal crayfish Pacifastacus leniusculus (Dana, 1852) in Croatia. J. Fish Dis. 2021, 1–16. [Google Scholar] [CrossRef]
- Dong, X.; Hu, T.; Liu, Q.; Li, C.; Sun, Y.; Wang, Y.; Shi, W.; Zhao, Q.; Huang, J. A Novel Hepe-Like Virus from Farmed Giant Freshwater Prawn Macrobrachium rosenbergii. Viruses 2020, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Lightner, D.V.; Pantoja, C.R.; Poulos, B.T.; Tang, K.F.J.; Redman, R.M.; Andrade, T.P.; Bonami, J.R. University Infectious Myonecrosis- New Disease in Pacific White Shrimp. Glob. Aquac. Advocate 2004, 7, 85. [Google Scholar]
- Edgerton, B.; Owens, L.; Glasson, B.; De Beer, S. Description of a small dsRNA virus from freshwater crayfish Cherax quadricarinatus. Dis. Aquat. Organ. 1994, 18, 63–69. [Google Scholar] [CrossRef]
- Sunarto, A.; Naim, S. Totiviruses of Crustaceans. In Aquaculture Virology; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 425–439. [Google Scholar]
- Zeng, D.; Chen, X.; Xie, D.; Zhao, Y.; Yang, C.; Li, Y.; Ma, N.; Peng, M.; Yang, Q.; Liao, Z.; et al. Transcriptome Analysis of Pacific White Shrimp (Litopenaeus vannamei) Hepatopancreas in Response to Taura Syndrome Virus (TSV) Experimental Infection. PLoS ONE 2013, 8, e57515. [Google Scholar] [CrossRef]
- Guo, Z.X.; He, J.G.; Xu, H.D.; Weng, S.P. Pathogenicity and complete genome sequence analysis of the mud crab dicistrovirus-1. Virus Res. 2013, 171, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, S.; Huang, J.; Wang, Q.; Li, K.; He, J.; He, J.; Weng, S.; Zhang, Q. Cryo-electron Microscopy Structures of Novel Viruses from Mud Crab Scylla paramamosain with Multiple Infections. J. Virol. 2019, 93, e02255-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldron, F.M.; Stone, G.N.; Obbard, D.J. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet. 2020, 16, e1007533. [Google Scholar] [CrossRef]
- Argenta, F.F.; Hepojoki, J.; Smura, T.; Szirovicza, L.; Hammerschmitt, M.E.; Driemeier, D.; Kipar, A.; Hetzel, U. Identification of Reptarenaviruses, Hartmaniviruses, and a Novel Chuvirus in Captive Native Brazilian Boa Constrictors with Boid Inclusion Body Disease. J. Virol. 2020, 94, e00001-20. [Google Scholar] [CrossRef] [PubMed]
- Dezordi, F.Z.; dos Santos Vasconcelos, C.R.; Rezende, A.M.; Wallau, G.L. In and Outs of Chuviridae Endogenous Viral Elements: Origin of a Potentially New Retrovirus and Signature of Ancient and Ongoing Arms Race in Mosquito Genomes. Front. Genet. 2020, 11, 1291. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; May, R.M. Population biology of infectious diseases: Part I. Nature 1979, 280, 361–367. [Google Scholar] [CrossRef]
- Heesterbeek, J.A.P. A brief history of R0 and a recipe for its calculation. Acta Biotheor. 2002, 50, 189–204. [Google Scholar] [CrossRef]
- Lloyd-Smith, J.O.; Schreiber, S.J.; Kopp, P.E.; Getz, W.M. Superspreading and the effect of individual variation on disease emergence. Nature 2005, 438, 355–359. [Google Scholar] [CrossRef]
- Lindenfors, P.; Nunn, C.L.; Jones, K.E.; Cunningham, A.A.; Sechrest, W.; Gittleman, J.L. Parasite species richness in carnivores: Effects of host body mass, latitude, geographical range and population density. Glob. Ecol. Biogeogr. 2007, 16, 496–509. [Google Scholar] [CrossRef]
- Gay, N.; Olival, K.J.; Bumrungsri, S.; Siriaroonrat, B.; Bourgarel, M.; Morand, S. Parasite and viral species richness of Southeast Asian bats: Fragmentation of area distribution matters. Int. J. Parasitol. Parasites Wildl. 2014, 3, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Webber, Q.M.R.; Fletcher, Q.E.; Willis, C.K.R. Viral Richness is Positively Related to Group Size, but Not Mating System, in Bats. Ecohealth 2017, 14, 652–661. [Google Scholar] [CrossRef]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2, vew022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, A.B.; Davies, T.J. Cross-species pathogen transmission and disease emergence in primates. Ecohealth 2009, 6, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; dos Santos Tavares, C.P.; Schott, E.J. Diversity and classification of reoviruses in crustaceans: A proposal. J. Invertebr. Pathol. 2021, 182, 107568. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.; Brodin, T.; Fogarty, S.; Sih, A. Non-random dispersal mediates invader impacts on the invertebrate community. J. Anim. Ecol. 2017, 86, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Conte, F.; Voslarova, E.; Vecerek, V.; Elwood, R.W.; Coluccio, P.; Pugliese, M.; Passantino, A. Humane slaughter of edible decapod crustaceans. Animals 2021, 11, 1089. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Reads after Trimming | Accession Number (SRA *) | % of Classified Reads (Diamond) | % of Reads Class. as Viral (Diamond) | % of Reads Class. as Riboviria (Diamond) | No. of Viral Read Class. as Riboviria (Diamond) |
|---|---|---|---|---|---|---|---|
| UF | 164,043,698 | 160,643,914 | SAMN20800941 | 15.47 | 0.00159 | 0.00094 | 1511 |
| UC | 149,357,758 | 146,166,391 | SAMN20800942 | 19.06 | 0.00895 | 0.00816 | 11,929 |
| DC | 100,131,420 | 97,563,944 | SAMN20800943 | 23.24 | 0.01737 | 0.01620 | 15,802 |
| DF | 136,134,398 | 132,904,626 | SAMN20800944 | 15.29 | 0.00961 | 0.00821 | 10,905 |
| Contig Name | Virus Name | GenBank Accesion | Contig Length | Blastx Results | ||
|---|---|---|---|---|---|---|
| Closest Protein Hit Name and Species | e-Value | Identities * | ||||
| Virus-like contig 4 | Signal crayfish associated reo-like virus 1 | OK317706 | 4234 | RdRp [Cherax quadricarinatus reovirus] | 2.74 × 10−20 | 106/422 (25%) |
| Virus-like contig 139 | Signal crayfish associated hepe-like virus 1 | OK317707 | 10,400 | hypothetical protein [Beihai hepe-like virus 4] | 2.64 × 10−117 | 213/480 (44%) |
| Virus-like contig 141 | Signal crayfish associated toti-like virus 1 | OK317708 | 8576 | hypothetical protein 4 [Wenzhou crab virus 5] | <1.00 × 10−250 | 353/869 (41%) |
| Virus-like contig 1 | Signal crayfish associated picorna-like virus 1 | OK317711 | 4587 | hypothetical protein [Beihai picorna-like virus 99] | 1.28 × 10−86 | 324/1251 (26%) |
| Virus-like contig 10 | Signal crayfish associated picorna-like virus 2 | OK317712 | 531 | hypothetical protein [Beihai picorna-like virus 99] | 2.75 × 10−39 | 79/175 (45%) |
| Virus-like contig 11 | Signal crayfish associated picorna-like virus 3 | OK317713 | 320 | hypothetical protein [Beihai picorna-like virus 99] | 4.97 × 10−51 | 80/105 (76%) |
| Virus-like contig 15 | Signal crayfish associated picorna-like virus 4 | OK317714 | 898 | hypothetical protein 1 [Changjiang picorna-like virus 6] | 1.67 × 10−37 | 85/151 (56%) |
| Virus-like contig 9 | Signal crayfish associated picorna-like virus 5 | OK317715 | 771 | hypothetical protein [Beihai picorna-like virus 99] Y | 1.30 × 10−96 | 170/257 (66%) |
| Virus-like contig 5 | Signal crayfish associated picorna-like virus 6 | OK317716 | 714 | hypothetical protein [Wenzhou picorna-like virus 38] | 1.56 × 10−19 | 62/152 (41%) |
| Virus-like contig 13 | Signal crayfish associated picorna-like virus 7 | OK317717 | 1894 | hypothetical protein [Beihai sesarmid crab virus 2] | 9.08 × 10−99 | 226/642 (35%) |
| Virus-like contig 66 | Signal crayfish associated tombus-like virus 1 | OK317718 | 4504 | replicase [Caledonia beadlet anemone tombus-like virus 1] | <1.00 × 10−250 | 352/862 (41%) |
| Virus-like contig 84 | Signal crayfish associated tombus-like virus 2 | OK317719 | 2981 | replicase [Caledonia beadlet anemone tombus-like virus 1] | 3.00 × 10−114 | 246/628 (39%) |
| Virus-like contig 55 | Signal crayfish associated tombus-like virus 3 | OK317720 | 1425 | RdRp [Riboviria sp.] | 2.00 × 10−92 | 150/299 (50%) |
| Virus-like contig 35 | Signal crayfish associated tombus-like virus 4 | OK317721 | 665 | RdRp [Riboviria sp.] | 4.00 × 10−32 | 76/161 (47%) |
| Virus-like contig 24 | Signal crayfish associated tombus-like virus 5 | OK317722 | 538 | hypothetical protein 1 [Hubei tombus-like virus 16] | 3.98 × 10−04 | 43/99 (43%) |
| Virus-like contig 169 | Signal crayfish associated tombus-like virus 6 | OK317723 | 301 | hypothetical protein 2 [Hubei unio douglasiae virus 2] | 2.00 × 10−42 | 69/82 (84%) |
| Virus-like contig 140 | Signal crayfish associated chu-like virus 1 | OK317724 | 2216 | hypothetical protein 2 [Beihai hermit crab virus 3] | 5.71 × 10−56 | 161/588 (27%) |
| Virus-like contig 65 | Signal crayfish associated chu-like virus 2 | OK317725 | 746 | hypothetical protein 2 [Beihai hermit crab virus 3] | 6.45 × 10−35 | 69/167 (41%) |
| Virus-like contig 7 | Signal crayfish associated chu-like virus 3 | OK317726 | 493 | RdRp [Beihai hermit crab virus 3] | 3.98 × 10−48 | 44/54 (81%) |
| Virus-like contig 145 | Signal crayfish associated chu-like virus 4 | OK317727 | 1009 | RdRp [Beihai hermit crab virus 3] | 1.63E × 10−60 | 120/219 (55%) |
| Virus-like contig 146 | Signal crayfish associated chu-like virus 5 | OK317728 | 418 | RdRp [Beihai hermit crab virus 3] | 1.00E × 10−27 | 54/98 (55%) |
| Virus-like contig 222 | Signal crayfish associated chu-like virus 6 | OK317729 | 356 | RdRp [Beihai hermit crab virus 3] | 2.00E × 10−45 | 80/111 (72%) |
| Virus-like contig 27 | Signal crayfish associated partiti-like virus 1 | OK317730 | 1188 | RdRp [Caledonia partiti-like virus] | 1.00E × 10−142 | 221/394 (56%) |
| Virus-like contig 116 | Signal crayfish associated picorna-like virus 8 | OK317731 | 734 | polyprotein [Picornaviridae sp.] | 3.70E × 10−10 | 42/88 (48%) |
| Virus-like contig 2 | Signal crayfish associated picorna-like virus 9 | OK317732 | 412 | hypothetical protein 1 [Picornavirales sp.] | 1.05E × 10−24 | 45/100 (45%) |
| Virus-like contig 36 | Signal crayfish associated picorna-like virus 10 | OK317733 | 457 | RdRp [Picornavirales sp.] | 4.32 × 10−16 | 60/102 (58%) |
| Virus-like contig 83 | Signal crayfish associated sobemo-like virus 1 | OK317734 | 431 | hypothetical protein [Hubei sobemo-like virus 43] | 1.00 × 10−50 | 84/143 (59%) |
| Virus-like contig 147 | Signal crayfish associated sobemo-like virus 2 | OK317709 | 512 | hypothetical protein 1 [Beihai sobemo-like virus 17] | 1.00E × 10−10 | 44/128 (34%) |
| Virus-like contig 30 | Signal crayfish associated narna-like virus 1 | OK317710 | 378 | RdRp [Beihai narna-like virus 18] | 5.60 × 10−18 | 48/128 (38%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bačnik, K.; Kutnjak, D.; Černi, S.; Bielen, A.; Hudina, S. Virome Analysis of Signal Crayfish (Pacifastacus leniusculus) along Its Invasion Range Reveals Diverse and Divergent RNA Viruses. Viruses 2021, 13, 2259. https://doi.org/10.3390/v13112259
Bačnik K, Kutnjak D, Černi S, Bielen A, Hudina S. Virome Analysis of Signal Crayfish (Pacifastacus leniusculus) along Its Invasion Range Reveals Diverse and Divergent RNA Viruses. Viruses. 2021; 13(11):2259. https://doi.org/10.3390/v13112259
Chicago/Turabian StyleBačnik, Katarina, Denis Kutnjak, Silvija Černi, Ana Bielen, and Sandra Hudina. 2021. "Virome Analysis of Signal Crayfish (Pacifastacus leniusculus) along Its Invasion Range Reveals Diverse and Divergent RNA Viruses" Viruses 13, no. 11: 2259. https://doi.org/10.3390/v13112259