









-
The family Orchidaceae is one of the most diverse and widespread plants, comprising approximately 28,000 species with 700 genera, accounting for roughly 9% of all vascular plants, growing in an extensive range of habitats but absent in the polar regions and deserts[1,2], as well as becoming a flagship taxon that has research significance in evolutionary biology. Orchids exhibit a high ornamental value and are favoured by many people, particularly horticulturists. The top five ornamental genera of orchids are as follows: Cattleya, Cymbidium, Dendrobium, Paphiopedilum and Phalaenopsis. Among them, Cymbidium has 71 species divided into three subgenera (Cymbidium, Cyperorchis, and Jensoa) and distributed in tropical and subtropical Asia and further south to Papua New Guinea and Northern Australia[3,4]. Some species of Cymbidium are widely cultivated, and some hybrids with important commercial value have been produced for over a hundred years[5]. China has a long-standing tradition of Cymbidium cultivation and appreciation, which is credited to have begun as early as the Tang Dynasty, or possibly as far back as the Confucius period. During these times, ornamental orchids were divided into two types: one scape with one flower as 'Lan' (兰), while one scape with multiple flower as 'Hui' (蕙). Nowadays, some species of Cymbidium are named Guolan (Chinese Orchid), including Chunlan C. goeringii, Jianlan C. ensifolium, Huilan C. faberi, Hanlan C. kanran, Molan C. sinense, and Lianbanlan C. tortisepalum, which have important humanistic value and have been cultivated as ornamental plants for many centuries. Among these Chinese orchids, C. goeringii represents a typical species of Cymbidium with a floral shape inclined to mutation, diverse floral colour, variable floral scent, and a long flowering period (Fig. 1). Over the past century, some wild species have been selected for breeding, such as 'Song Mei' and 'Jin Yuan Die'. However, the lack of high-quality genomic data limits the study of the evolution and cultivation application of C. goeringii.

Figure 1.
A flowering plant of Cymbidium goeringii 'Da Fu Gui'.
To improve our understanding of the molecular mechanism of morphological traits in Cymbidium, the genome of C. goeringii was sequenced and analysed. Genomic analysis and comparison with other sequenced orchids could yield new insights into the key innovations in the evolution of C. goeringii and provide important genomic data for the cultivation of Cymbidium.
-
The 19-mer analysis of C. goeringii (2N = 2X = 40)[6] showed that the C. goeringii genome size was approximately 4.35 Gb with heterozygosity of 2.73% (Supplemental Fig. S1 & Supplemental Table S1). PacBio sequencing was performed to assemble the C. goeringii genome to the contig level. PacBio completed the sequencing of four cells and obtained a total of 477.90 Gb raw data (Supplemental Table S2). The assembled genome size was 4.10 Gb with a corresponding contig N50 value of 1.04 Mb (Supplemental Table S3). In addition, the Benchmarking Universal Single-Copy Orthologues (BUSCO) showed that the completeness of the assembled genome was 86.90% (Supplemental Table S4), indicating that the C. goeringii genome assembly was relatively complete and of high quality. High-throughput chromosome conformation capture (Hi-C) was performed to assemble the genome to the chromosome level and 296.04 Gb raw reads were obtained (Supplemental Table S5). A total of 4.07 Gb sequences (95.86%) were mapped to 20 pseudochromosomes (Supplemental Table S6 & S7). The lengths of the pseudochromosomes ranged from 88.08−280.68 Mb, with an N50 value of 209.04 Mb (Supplemental Table S6 & S7). The chromatin interaction data suggested the high quality of our Hi-C assembly (Supplemental Fig. S2).
We estimated 77.65% of the repetitive sequences in the C. goeringii genome (Supplemental Fig S3, S4 & Supplemental Table S8). Transposable elements (TEs) were the main component (75.78%), with the long terminal repeats (LTRs) family being the largest part (62.02%) of these transposons (Supplemental Table S9). Of the protein-coding genes, 30,897 were confidently annotated in the C. goeringii genome (Supplemental Table S10 & S11). BUSCO assessment indicated that the completeness of the annotated genome was 91.00% (Supplemental Table S12). In addition, 147 microRNAs, 493 transfer RNAs, 1,544 ribosomal RNAs, and 528 small nuclear RNAs were identified in the C. goeringii genome (Supplemental Table S13). Also, 29,272 genes (94.74%) were predicted to be annotated to functional databases, among which 21,930 and 21,763 were annotated to Kyoto Encyclopaedia of Genes and Genomes (KEGG) terms and Clusters of Orthologous Groups for Eukaryotic Complete Genomes (KOG), respectively (Supplemental Table S14 & Supplemental Fig. S5−S8).
Phylogenomic and gene family evolution analyses
-
To infer the phylogenetic position of C. goeringii, a phylogenomic analysis was performed using 267 single-copy gene families extracted from 17 different plant species (Supplemental Fig. S9 & Supplemental Table S15). The result showed that C. goeringii is a sister to P. equestris and forms a clade with D. catenatum and G. elata in the Epidendroideae (Supplemental Fig. S9). The estimated Orchidaceae divergence time was 121.96 Mya; the divergence time between Apostasioideae and subfamily Epidendroideae was 82.44 Mya. The divergence time between C. goeringii and P. equestris was 38.08 Mya (Fig. 2a).
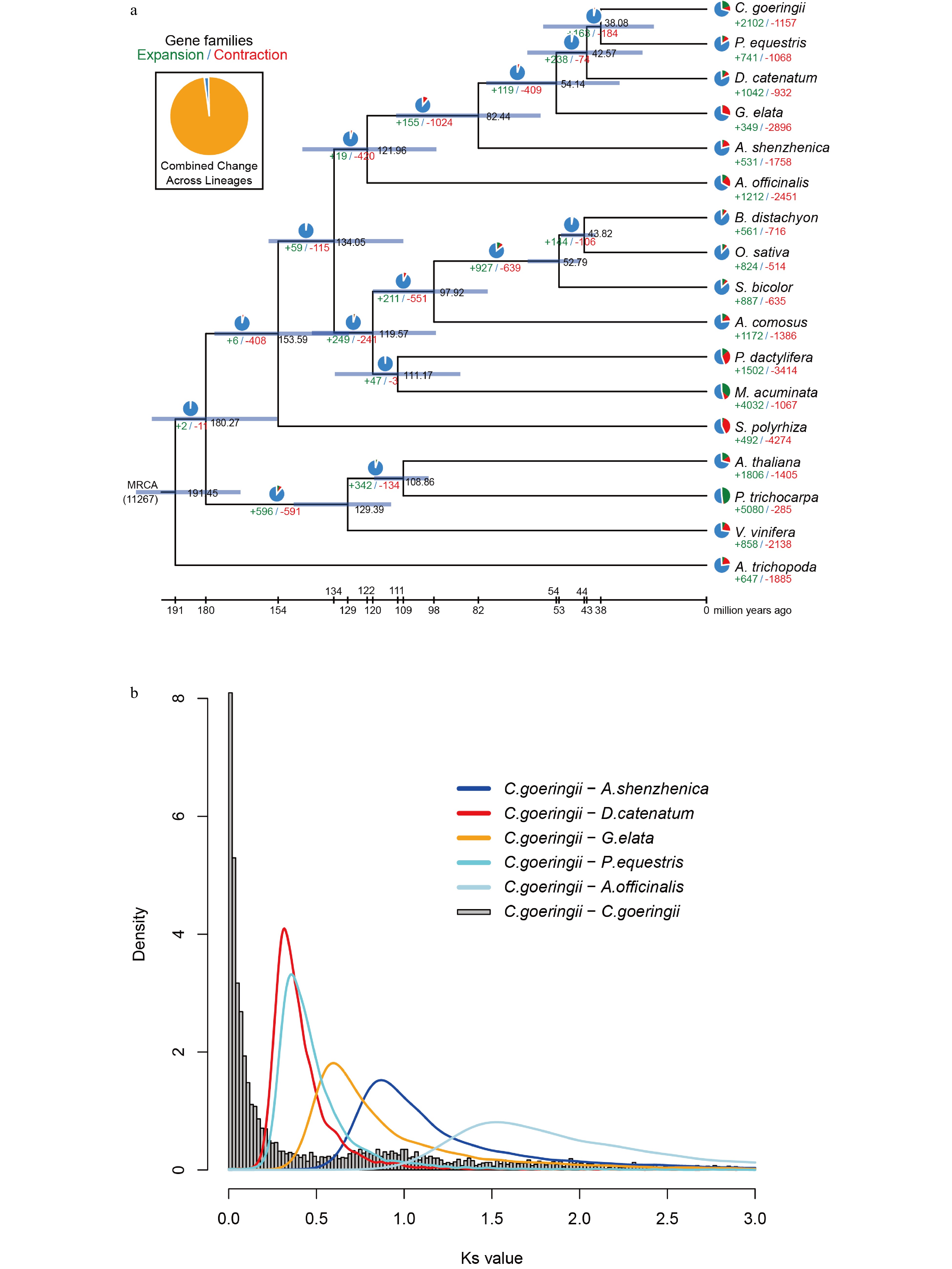
Figure 2.
Gene family evolution and whole-genome duplication of the C. goeringii genome. (a) The expansion and contraction of gene families and phylogenetic relationships and divergence times between C. goeringii and other plant species. The numbers in green represent the number of expanded gene families, and the numbers in red represent the number of contracted gene families. The blue colour in the circle indicates the gene families with a constant copy number. (b) Ks distribution of C. goeringii. C. goeringii showed two peaks at 0.8−1.0 and 1.7, indicating that C. goeringii experienced a τ event and a WGD event shared with the other extant orchids.
We also investigated gene family evolution of Orchidaceae. Gene family expansion and contraction showed that 155 gene families were expanded in the lineage leading to Orchidaceae, whereas 1,024 gene families were contracted (Fig. 2a). In the C. goeringii genome, 2,102 and 1,157 gene families were expanded and contracted, respectively, in which there were 186 gene families with 1,921 genes significantly expanded, and 21 gene families with 10 genes significantly contracted (Fig. 2a). C. goeringii had more expanded gene families than other sequenced orchids[7−10]. Enrichment analysis indicated that significantly expanded gene families of C. goeringii were especially enriched in the GO terms 'transporter activity', 'transmembrane transporter activity', and 'positive regulation of biological process', and in the KEGG pathways 'metabolic pathways', 'biosynthesis of secondary metabolites', 'starch and sucrose metabolism', and 'fatty acid elongation' (Supplemental Table S16 & S17). In addition, genome comparative analysis showed that the C. goeringii genome is composed of 1,301 unique gene families with 2,803 genes (Supplemental Table S15). The unique gene families were significantly enriched in the GO terms 'nuclear-transcribed mRNA catabolic process', 'nonsense-mediated decay cellular nitrogen', and 'compound catabolic process', and in the KEGG pathways 'diterpenoid biosynthesis' and 'biosynthesis of ansamycins' (Supplemental Table S18 & S19).
Whole-genome duplication
-
Whole-genome duplication (WGD) events are an important feature in many taxa, it is also an efficient way to expand the genome size[11]. The protein sequences of P. equestris, P. aphrodite, and D. catenatum were analysed to obtain the gene pairs in the collinear region for the distributions of synonymous substitutions per synonymous site (Ks) analysis. The collinearity of C. goeringii and P. equestris showed that the chromosomes had a good one-to-one correspondence (Supplemental Fig. S10). The Ks values of C. goeringii, P. aphrodite, P. equestris, and D. catenatum were further estimated to more precisely infer the WGD of C. goeringii.
The distributions of Ks for paralogous C. goeringii genes showed two peaks at Ks = 0.8 – 1.0 and at Ks = 1.7 (Fig. 2b), implying that two WGD events occurred in the C. goeringii genome. The Ks differentiation peaks of C. goeringii–A. officinalis and C. goeringii–A. shenzhenica were located between the values of the two Ks peaks in the C. goeringii genome (Fig. 2b), suggesting that the common ancestor of A. officinalis, A. shenzhenica, and C. goeringii experienced an older WGD event before they diverged. The Ks differentiation peaks of C. goeringii–G. elata, C. goeringii–D. catenatum, and C. goeringii–P. equestris were all smaller than the values of the two Ks peaks in the C. goeringii genome (Fig. 2b), suggesting that the common ancestor of G. elata, D. catenatum, P. equestris, and C. goeringii experienced two WGD events. Previous studies reported that the genomes of A. shenzhenica[7], G. elata[8], D. catenatum[9], P. equestris[10], C. ensifolium[12] and C. sinense[13] experienced two WGD events. The older one is a τ event shared by most monocots, and the most recent is a WGD event shared by orchid ancestors[7]. In summary, it was inferred that the C. goeringii genome experienced a τ event and a WGD event shared with the other extant orchids without having an independent WGD event.
MADS-box genes and organs development in C. goeringii
-
MADs-box family genes are important transcriptional regulators and play a key role in plant growth and development[7, 14−16]. In the present study, the MADS-box genes were identified from the C. goeringii genome to allow a comprehensive understanding of the molecular mechanism of organ development in C. goeringii. A total of 74 MADS-box genes were identified in C. goeringii (Table 1 & Supplemental Table S20). The number of MADS-box genes in C. goeringii were higher than were found in A. shenzhenica (36), C. ensifolium (71), D. catenatum (63), and P. equestris (51)[7,9,10]. C. goeringii had 44 type II MADS-box genes, which was higher than was found in P. equestris (29), A. shenzhenica (27), D. catenatum (35) and C. ensifolium (38) (Supplemental Fig. S11 & Table 1). We found that the MADS-box gene subfamily B-AP3 and E-classes were reduced in A. shenzhenica (two B-AP3 and three E-class genes), compared with four B-AP3 and six E-class genes in C. goeringii: this is consistent with a previous study that reported the lower gene numbers of B-AP3 and E-classes genes represent an ancestral state, responsible for producing the actinomorphic flower, and the higher numbers of these two genes can produce bisymmetry flowers[7]. Transcriptome analysis showed that B-AP3 and E-class genes were mainly expressed in the floral organs (petals, sepals, lips, and column), with low or no expression in the vegetative organs (root, stem, and leaf) of C. goeringii, indicating that these two MADS-box subfamilies mainly relate to the growth and development of floral organs in C. goeringii (Supplemental Fig. S12). In the C. goeringii genome, we did not find any type I Mβ MADS-box genes, consistent with the lack of type I Mβ MADS-box genes resulting in the lack of endosperm seen in orchids[7] (Supplemental Fig. S11). Two SVP genes (GL09236 and GL14819) were highly expressed in the roots, stem and leaves, this expression pattern was the same as Arabidopsis[17] (Supplemental Fig. S12), suggesting that the SVP gene may be related to the growth and development of vegetative organs in C. goeringii. The absence of the AGL12 gene and the contraction of the ANR1 gene indicated that C. goeringii may be an epiphytic orchid without 'true' terrestrial growth.
Table 1. MADS gene family of five orchid species.
Category A. shenzhenica[7] P. equestris[10] D. catenatum[9] C. ensifolium[12] C. goeringii* Type II (Total) 27 29 35 38 44 MIKCc 25 28 32 34 38 A 2 3 4 4 4 AGL6 2 3 3 3 4 AGL12 1 0 0 0 0 AGL15 0 0 0 0 0 ANR1 4 2 3 1 1 AP3 2 4 4 4 4 B-PI 1 1 1 1 1 Bs 1 1 2 7 1 C/D 4 5 4 4 4 E 3 6 5 4 6 FLC 0 0 0 0 0 OsMADS32 1 0 1 1 1 SOC1 2 2 2 3 4 SVP 2 1 3 2 4 MIKC* 2 1 3 4 6 Type I (Total) 9 22 28 33 30 Mα 5 10 15 27 26 Mβ 0 0 0 0 0 Mγ 4 12 13 6 4 Total 36 51 63 71 74 * This study. Expression and epigenetic regulation of MADS-box genes
-
Previous studies have shown that expanded B-AP3 and E-clades with members that have different expression patterns in floral organs associated with the innovation of the labellum (lip) and gynostemium (column) in orchids[7]. We found in C. goeringii, the lip formation is controlled by AGL6-3, BAP3-1, and BAP3-4, and the C-class genes are mainly expressed in the columns and related to their formation (Supplemental Fig. S12 & Supplemental Table S20). Normal flower development is consistent with the Homeotic Orchid Tepal (HOT) model[16]. There are flowers with lip-and column-like sepal or petal in wild populations of C. goeringii. Therefore, we analysed the regulation mechanisms of MADS-box genes in mutant flowers of C. goeringii.
Lip-like petal mutant ( 蕊蝶花, Ruidiehua)
-
In this mutant, the petals were mutated to the structure of a lip, forming a three-lip flower without petals (Fig. 3b). Compared to those in normal petals (Fig. 3a), one AGL6 gene (GL08067) and one AP3 gene (GL11355) were highly expressed in lip-like petals, while GL08067 was not expressed in normal petals (Supplemental Fig. S13), suggesting that the genes regulating the lips have occupied the expression position of genes involved in petal development, the expression of the latter being inhibited, leading to lip-like petals.

Figure 3.
Normal flower morphology and mutants. (a) Normal flower; (b) lip-like petal mutant; (c) lip-like sepal mutant; (d) column-like petal mutant.
Lip-like sepal mutant ( 蝶花, Diehua)
-
Lateral sepals of flowers become a semi-lip structure, forming a lip-like sepal mutant, similar to a butterfly, namely 'Diehua' (Fig. 3c). Transcriptomic analysis showed that the expression of three B-AP3 genes (GL11355, GL29814, and GL17940) in lip-like sepals compared to normal sepals increased significantly. Meanwhile, the expression of one E gene (GL23742) was highly expressed in the normal sepals, while it had low expression in the lip-like sepals (Supplemental Fig. S13). In conclusion, we suggest that the genes regulating lip formation have occupied the expression position of genes involved in sepal development, whose expression were also being inhibited, forming a lip-like sepal mutant.
Column-like petal mutant ( 梅瓣花, Meibanhua)
-
In this mutant, the petals of flowers mutate into a shape similar to a column (Fig. 3d). Transcriptomic analysis showed that the expression of one C-class gene (GL11898) was highly expressed in column-like petals, while one E gene (GL23740) was significantly decreased (Supplemental Fig. S13), similar to the genes that regulate column development. It is suggested that the expression position and expression of genes involved in petal development has been occupied and inhibited by genes regulating column development, respectively, contributing to column-like petals.
Tepal-like leaves
-
The terminal leaves of plant become the tepal-like structure in that mutant. Transcriptomic analysis showed that the expression of MADS-box genes that are closely related to the development of floral organs was significantly increased in tepal-like leaves, in comparison to normal leaves (Supplemental Fig. S14), indicating that the increased expression of MADS-box genes related to flower development in leaves led them to change into tepal-like leaves.
Herein, we suggested that floral organ development in orchids is not limited to the classical HOT model[16] and ABCDE[18] model. Genes controlling sepal, petal, lip and column can occupy each other's expression position thus inhibiting or repressing the other's expression, forming a variety of mutants that differ from normal flowers. It is possible that changes in plant hormones may lead to the abnormal expression of these genes, particularly the metabolism of gibberellin (GA). We noticed that GA was commonly used to increase the number of scape of orchids in the orchid industry, which often yielded these types of mutant flowers, whereas the specific regulation mechanism requires further research.
Floral colour regulatory pathway in C. goeringii
-
C. goeringii is popular worldwide for their variety of colors. Carotenoids and anthocyanins are the main flower pigments. Pale-yellow with purple-red spots, green-yellow, and purple-red flowers from three C. goeringii varieties were used to explore the floral colour regulatory pathway in C. goeringii.
Carotenoids are the most widely distributed pigment in nature and can be divided into two categories: carotene and lutein[19]. Carotenoids generally show bright reds, oranges, and yellows as they mainly absorb short-wavelength light[20]. The expression levels of genes involved in the carotenoid biosynthesis pathway of C. goeringii varieties were determined using transcriptome analysis. The results showed that PDS was highly expressed in the sepals of pale-yellow with purple-red spots and green-yellow flowers (Fig. 4), and was highly expressed in the petals of pale-yellow with purple-red spots flowers. ZDS was highly expressed in the sepals, petals, and lips of pale-yellow flowers with purple-red spots and purple-red flowers. CRTISO was highly expressed in the sepals, petals, and lips of pale-yellow flowers with purple-red spots. LCYE was highly expressed in the sepals and petals of green-yellow flowers, and was highly expressed in the petals of purple-red flowers. LCYB was highly expressed in the sepals, petals, and lips of pale-yellow flowers with purple-red spots and green-yellow flowers (Fig. 4). ZEP was highly expressed in the sepals, petals, and lips of pale-yellow flowers with purple-red spots and green-yellow flowers, and was highly expressed in the petals of purple-red flowers. BCH was highly expressed in the sepals and petals of green-yellow flowers. Our results suggest that the high expression of BCH, LCYE, LCYB, CRTISO and PDS might be positive activators and can prompt carotenoid accumulation of pale-yellow with purple-red spots and green-yellow flowers.
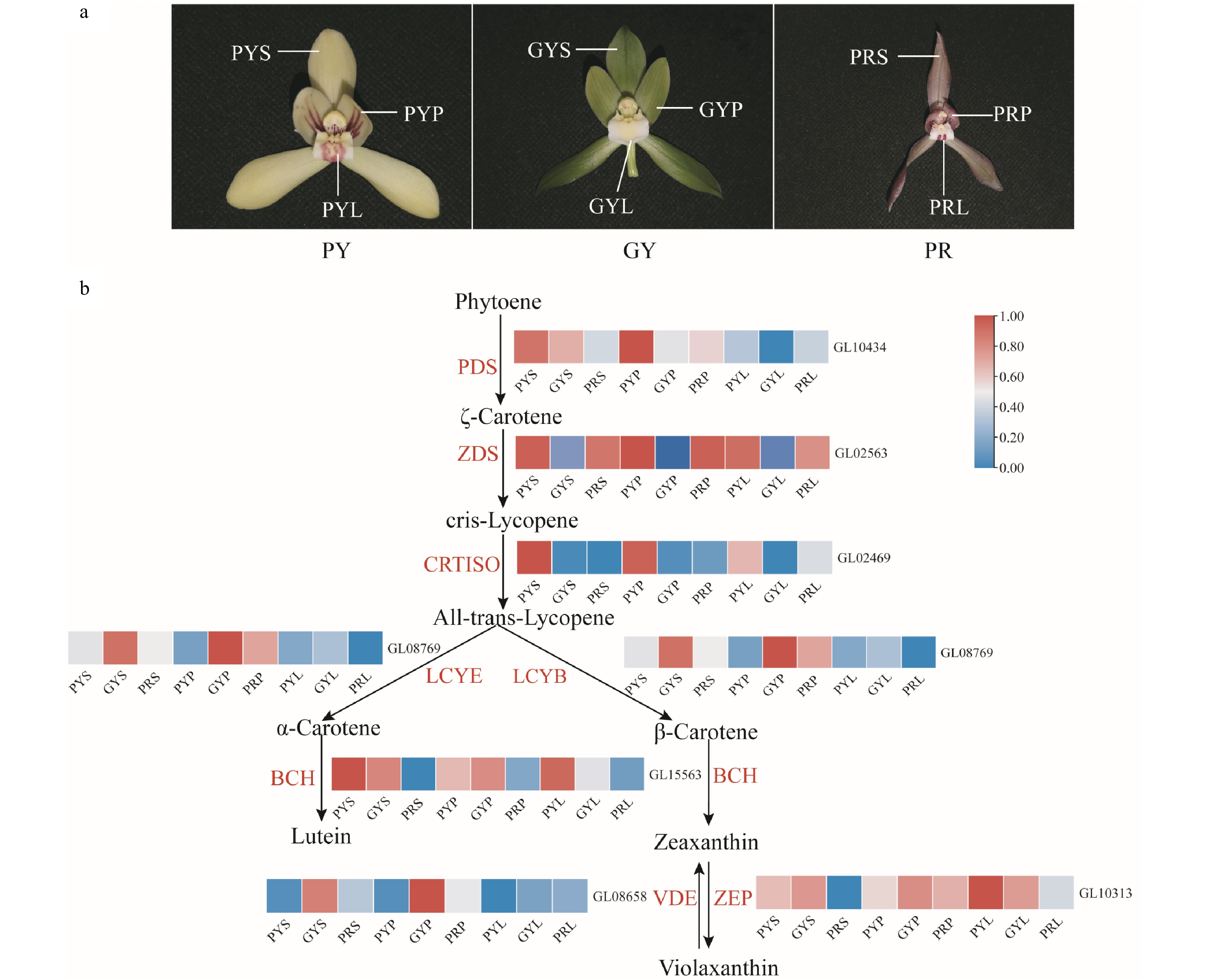
Figure 4.
Expression regulation of carotenoid metabolic pathway-related genes involved in flower colours in C. goeringii. (a) Three flower colour types. PY, pale-yellow flower with purple-red spots; GY, green-yellow flower; PR, purple-red flower. (b) The pathway of floral carotenoid biosynthesis. PYS, sepals of pale-yellow flowers with purple-red spots; PYP, petals of pale-yellow flowers with purple-red spots; PYL, lips of pale-yellow flowers with purple-red spots; GYS, sepals of green-yellow flower; GYP, petals of green-yellow flower; GYL, lips of green-yellow flower; PRS, sepals of purple-red flower; PRP, petals of purple-red flower; PRL, lips of purple-red flower. The heatmap was plotted from the FPKM value and performed with min-max normalisation. Red indicates high levels of expression, while blue indicates low levels of expression. The abbreviated names of enzymes (for full names see Supplemental Table S23) involved at each step are shown in red in each catalytic step[18].
Anthocyanins are the main components of flavonoids, showing a wide range of colours from pink to blue-purple, and play an irreplaceable role in the process of flower colour formation[21, 22]. Transcriptome analysis of C. goeringii showed that F3'H and F3'5'H were highly expressed in the sepals, petals, and lips of pale-yellow with purple-red spots, green-yellow and purple-red flowers (Fig. 5). ANS and UFGT were highly expressed in the sepals, petals, and lips of purple-red flowers (Fig. 5). Our results suggest that the high expression of ANS and UFGT can prompt anthocyanin accumulation in purple-red flowers. In flowers, R2R3-MYB transcription factors play an important role in regulating anthocyanin biosynthesis, especially members belonging to subgroup 6 (S6), such as AtMYB75, AtMYB90, AtMYB113, and AtMYB114 that control anthocyanin biosynthesis in Arabidopsis[23]. In Orchidaceae, three R2R3-MYB genes, PeMYB2, PeMYB11, and PeMYB12, of Phalaenopsis control the overall red, red spot, and texture pattern of the petals, respectively. PeMYB11 was responsive to the red spots in the callus of the lip, and PeMYB12 participated in full pigmentation in the central lobe of the lip[24]. In this study, six related genes of the seven R2R3-MYB genes were identified from the C. goeringii genome (Supplemental Fig. S15). In C. goeringii varieties, GL19121 was highly expressed in the lips of pale yellow flowers with purple-red spots (Supplemental Fig. S15), which suggested that GL19121 might be responsive to red spots in the lips of pale yellow flowers with purple-red spots. GL03052, GL16686, and GL12688 were highly expressed in the sepals and petals of purple-red flowers. GL13772 and GL18847 were highly expressed in the sepals, petals, and lips of purple-red flowers, which suggested that these six genes can prompt anthocyanin accumulation in C. goeringii purple-red flowers. In conclusion, the different expression levels of the genes related to carotenoid and anthocyanin pathways result in the various floral colours of C. goeringii.
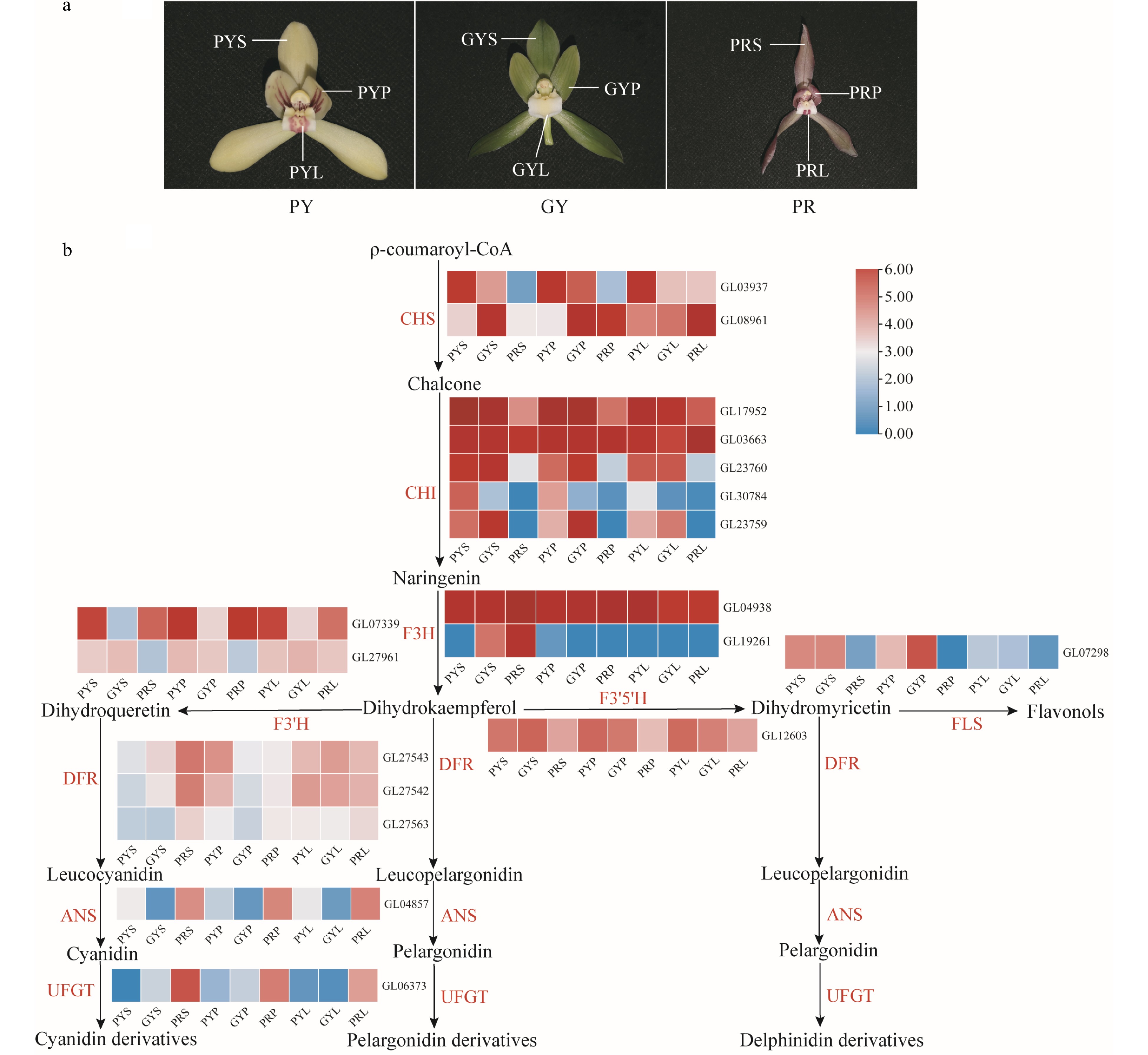
Figure 5.
Expression regulation of anthocyanin metabolic pathway-related genes involved in coloured flowers of C. goeringii. (a) Three flower colour types. PY, pale-yellow flower with purple-red spots; GY, green-yellow flower; PR, purple-red flower. (b). The pathway of floral anthocyanin biosynthesis. PYS, sepals of pale-yellow flower with purple-red spots; PYP, petals of pale-yellow flower with purple-red spots; PYL, lips of pale-yellow flower with purple-red spots; GYS, sepals of green-yellow flower; GYP, petals of green-yellow flower; GYL, lips of green-yellow flower; PRS, sepals of purple-red flower; PRP, petals of purple-red flower; PRL, lips of purple-red flower. The heatmap was plotted from the FPKM value and performed using min-max normalisation. The red indicates high levels of expression, while blue indicates low levels of expression. The abbreviated names of enzymes (for full names seeSupplemental Table S23) in each catalytic step[19] are shown in red.
Floral scent regulatory pathway in C. goeringii
-
Scent is an important property of flowers and is an important factor affecting the ornamental value of orchids[25]. Flower scent is composed of many kinds of volatile organic compounds, such as terpenes, styrene, benzene, fatty acids and their derivatives. In C. goeringii, floral scent compounds have been studied in various developmental stages during flowering, and terpenes are major compounds in the C. goeringii floral scent profile[25]. In this study, the transcriptome of C. goeringii flowers was generated at four different organs and three developmental stages, the comprehensive gene expression information in the whole genome will provide an understanding of the floral scent regulatory pathway in the C. goeringii.
For the cytosolic mevalonate (MVA) pathway, the gene HMGR (GL10633) was highly expressed in the sepals, petals, and lips of the full flowering stage in C. goeringii; three IDI genes were highly expressed in the sepals, petals, lips and column of the full flowering stage in C. goeringii, the same expression pattern as the FDPS gene (Fig. 6). In the plastidial methylerythritol phosphate (MEP) pathway, one DXS gene (GL19566) was highly expressed in the sepals, petals, lips and column of the full flowering stage, the same expression pattern as CMK (GL02895), HDS (GL21637), and HDR (GL22804 and GL22797) genes. Our results showed that most genes in the MVA pathway and MEP pathway increased expression during the flower development of C. goeringii, and had the strongest expression during the full flowering stage, suggesting the increase in scent volatiles during flower development. Also, the genes related to the MVA pathway and MEP pathway were mainly highly expressed in the sepals, petals, and lips, indicating that floral scent is mainly produced in the perianth of C. goeringii.
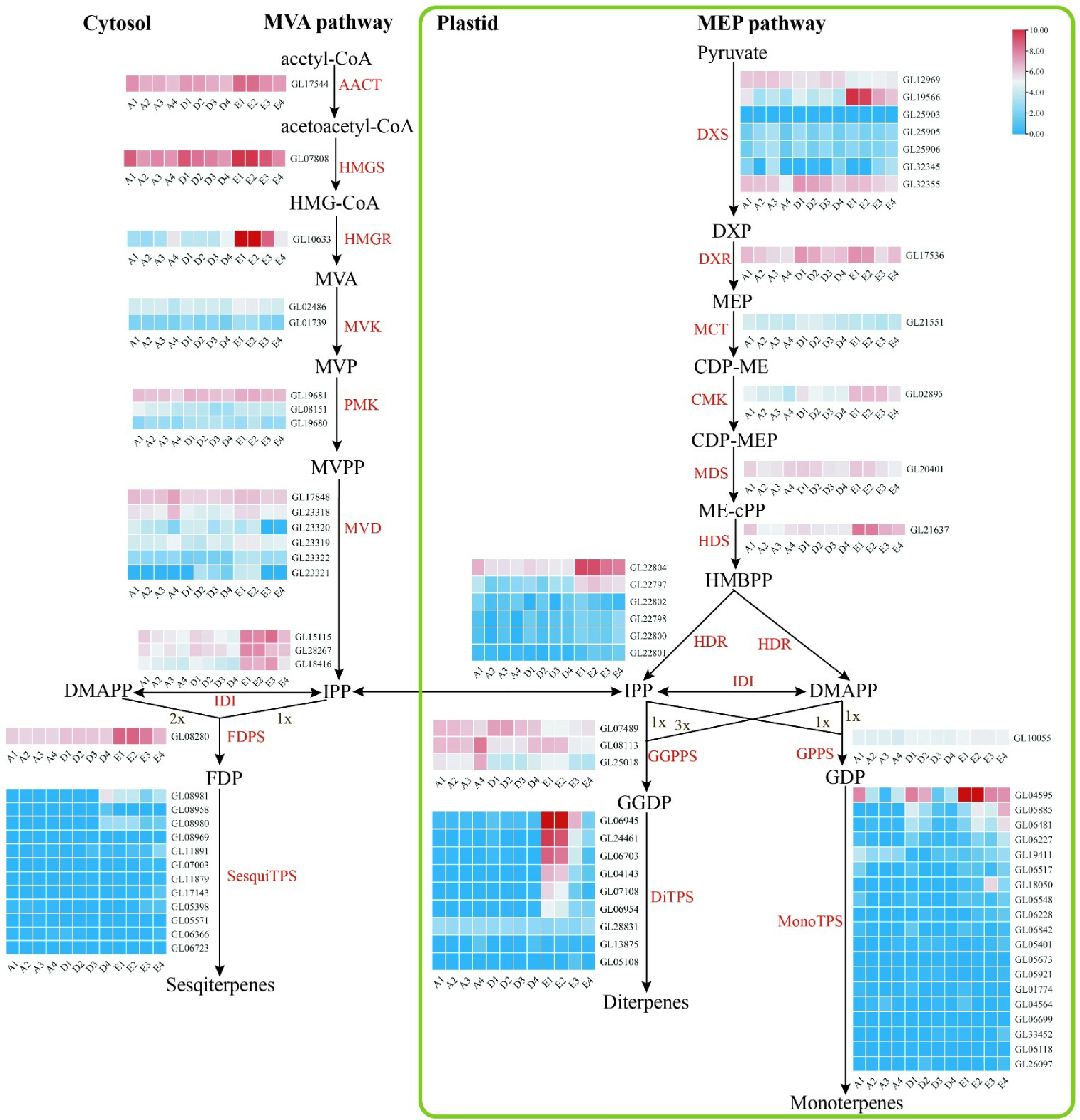
Figure 6.
Floral scent metabolic pathway and expression regulation of genes in C. goeringii. Tissue-specific relative expression profiles (red–blue scale) of genes implicated in terpenoid biosynthesis (heat map). Intermediates are shown in black, and the enzymes (for full names see Supplemental Table S23) involved in each step are shown in red. A1, sepals of 0.5−0.8 cm floral bud; A2, petals of 0.5−0.8 cm floral bud; A3, lips of 0.5−0.8 cm floral bud; A4, column of 0.5−0.8 cm floral bud; B1, sepals of 2−2.5 cm floral bud; B2, petals of 2−2.5 cm floral bud; B3, lips of 2−2.5 cm floral bud; B4, column of 2−2.5 cm floral bud; C1, sepals of blooming flower; C2, petals of blooming flower; C3, lips of blooming flower; C4, column of blooming flower. The abbreviated names of enzymes (for full names see Supplemental Table S23) in each catalytic step[24] are shown in red.
The terpene synthase gene (TPS) is a key gene that participates in the generation of terpenes[26]. Substantial activity of this enzyme has been associated with rapid accumulation of terpenes in plants. In the present study, 40 TPS gene family members were identified from C. goeringii and classified into four subfamilies based on phylogenetic analysis (Supplemental Fig. S16). The number of SPS genes in C. goeringii was more than that of D. catenatum (39 SPS genes), P. equestris (21 SPS genes)[10], and A. shenzhenica (six SPS genes). Four of these were highly expressed in the sepals, petals, and lips of blooming flowers, indicating that diterpenes and monoterpenes are mainly produced in the late stage of the perianth (Fig. 6). In conclusion, our results show that diterpenes and monoterpenes maybe the main compounds of C. goeringii, and are mainly produced in perianth at the full flowering stage.
Colourful leaf regulatory pathway in C. goeringii
-
Chlorophyll (chlorophyll, Chl) is an important pigment involved in photosynthesis in green plant chloroplasts, which plays an important role in energy capture and energy transfer for photosynthesis[27]. In general, leaf greening is mainly due to the absolute proportion of chlorophyll, while the formation of yellow leaves is mainly due to the degradation of chlorophyll, which makes the colour of carotenoids dominate the leaves[28,29]. Colourful leaves are an important ornamental characteristic of C. goeringii, which is known as 'line art' or 'leaf art', and have always been attractive to breeders and consumers. However, the formation mechanism of the 'arts' of C. goeringii is largely unknown. In this study, the leaf yellowing mechanism of C. goeringii in the genome and transcriptomes was studied from chlorophyll biosynthesis and degradation pathway genes. A total of 30 genes related to chlorophyll biosynthesis in the C. goeringii genome were identified. The expression of these 30 genes in the normal green leaves, yellow tissue, and green tissue was basically the same (Supplemental Fig. S17), which indicated that the chlorophyll synthesis pathway was not the cause of the leaf yellowing mechanism of C. goeringii. A total of eight genes related to chlorophyll degradation in the C. goeringii genome were identified, most of which showed different expression patterns in different mutants of C. goeringii, and most of them were expressed at higher levels in yellow tissue (Fig. 7), indicating that leaf yellowing is caused by chlorophyll breakdown that unmasks yellow pigments. The pheophorbide, an oxygenase gene (PAO), encodes a key enzyme of chlorophyll degradation; the expression of one homologous gene of PAO (GL02557) in yellow tissue was increased significantly compared to that in the normal green leaves and green tissue. Together, our study revealed that the high expression of genes related to chlorophyll degradation is the main reason for colourful leaves.
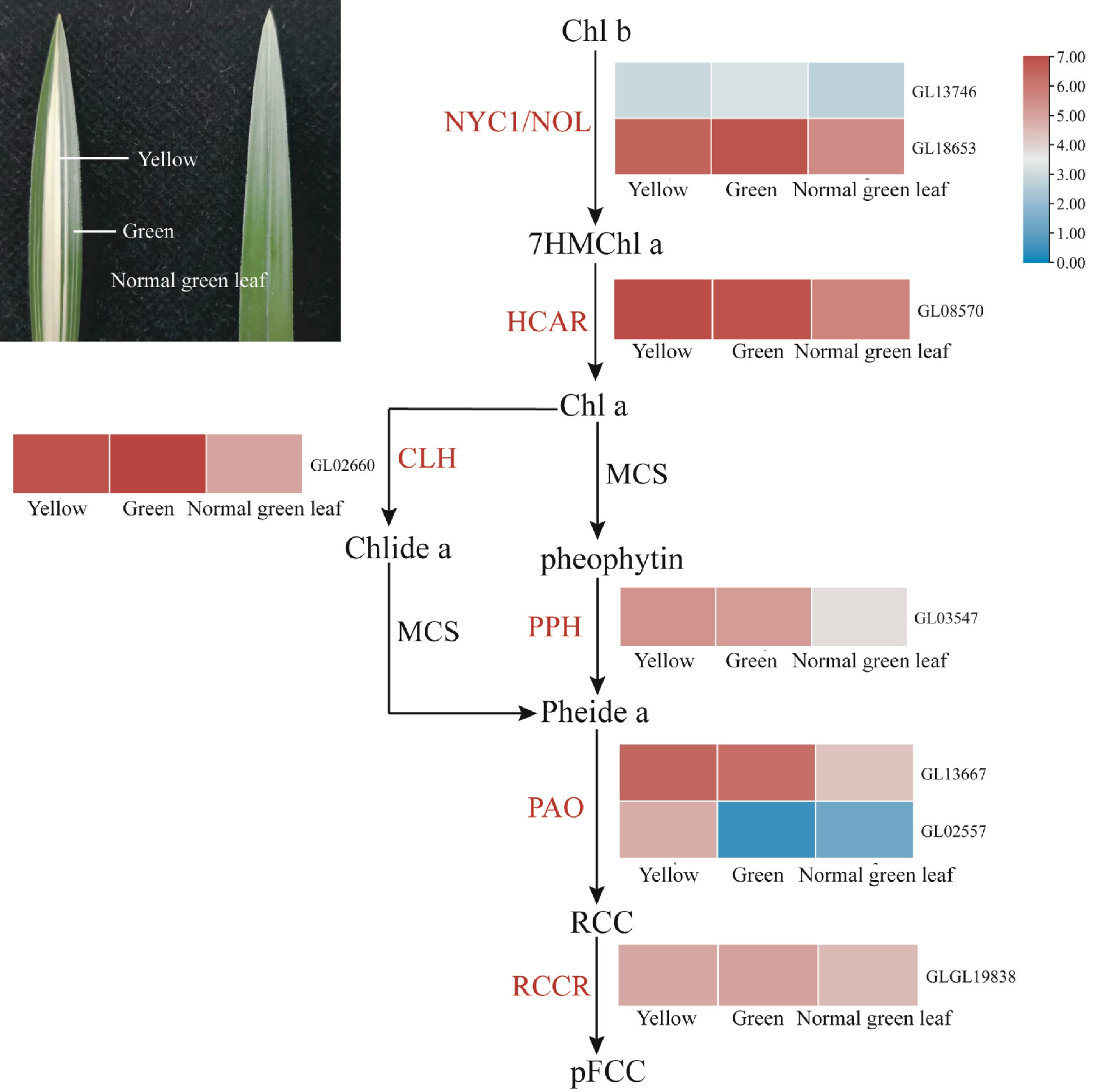
Figure 7.
Expression regulation of genes involved in chlorophyll degradation in coloured leaves of C. goeringii. Intermediates are shown in black, and the enzymes (for full names see Supplemental Table S23) involved at each step are shown in red. Normal green leaf, tissue of normal green leaf; Yellow, yellow tissue of yellow-green leaf mutant type; Green, green tissue of yellow-green leaf mutant type.
The resistance genes and adaptive evolution
Disease resistance genes
-
Plants have developed a variety of immune systems against the invasion of pests and diseases in the environment[30]. One of the most complex and effective immune systems is the recognition of specific pathogens, mediated by resistance genes. Resistance genes constitute a very large polygenic family, which has high polymorphism and diverse recognition characteristics[31]. According to the domain and function of the R gene, it can be divided into five types: nucleotide-binding site and leucine-reach repeats (NBS-LRR), receptor-like kinase (RLK), receptor-like protein (RLP), serine/theorine kinase (STK) genes and other genes that do not contain regular domains[32]. The R genes of orchids are mainly distributed in the NBS-LRR type. The genome of C. goeringii, D. catenatum, and P. equestris possess 83, 157 and 79 R genes (Supplemental Table S21), respectively[9]. The R gene family is related to resistance, the number of R gene family may be related to species adaptability and its distribution range. D. catenatum is the species most widely distributed, followed by C. goeringii and P. equestris, which is consistent with the number of R genes[10].
Heat-shock proteins
-
Heat shock proteins (Hsp) are a family of proteins produced by cells in response to exposure to stressful conditions in the environment[33]. Hsp genes are associated with stress caused by heat shock and other abiotic or biotic factors. According to the molecular weight, Hsp genes can be divided into small Hsps, Hsp20, Hsp40, Hsp60, Hsp70, Hsp90 and Hsp110[9]. Plants mainly include the gene families of Hsp20, Hsp70 and Hsp90. Hsp70 family members have organized protein aggregation, help inactive protein refolding, protein input and transport signal transduction and transcriptional activation[34]. The genome of C. goeringii, D. catenatum, and P. equestris possess 19, 20 and 9 Hsp70 genes (Supplemental Table S22). C. goeringii and D. catenatum are mainly distributed in subtropical and temperate regions of Asia. C. goeringii can adapt slightly to cold, conditions D. catenatum can adapt to drought or humidity, endure low temperature and high temperature[9], and both have a wider distribution range than P. equestris. More HSP70 gene family members may help C. goeringii and D. catenatum adapt to a variety of habitats, making their distribution wider than P. equestris.
-
As an important ornamental plant with high cultural value in Asia, the genome of a typical Guolan C. goeringii was sequenced and analysed. The genome of C. goeringii revealed two WGD events: a recent event shared by all orchids, not only itself, and an older event shared by most monocots (τ event). MADS-box genes were analysed in C. goeringii to reveal key genes regulating floral organs in normal flowers and mutants. The results suggested that the occupying expression of genes involved in floral organ development of normal flowers leads to the formation of mutants. The variety of colours seen in C. goeringii is caused by the different expression levels of anthocyanin metabolism-related, carotenoid metabolism-related, and R2R3-MYB genes, showing that the increased expression levels of genes related to chlorophyll degradation pathways led to the formation of colourful leaves. The genes of floral scent biosynthesis in C. goeringii were identified. Floral scent regulation mechanism analysis showed that diterpenes and monoterpenes maybe the main compounds of C. goeringii, and are mostly produced in the perianth at the full flower stage. We also analysed the resistance genes and revealed the relationship with the adaptative evolution of C. goeringii. Our results provide insight into the molecular mechanisms of orchid-specialised floral organs, floral scent, colours, and adaptive evolution.
-
DNA was extracted from young leaves of C. goeringii with CTAB reagents[35]. The RNA Plant Plus Kit (Tiangen, DP473) was used to extract the RNA from roots, pseudobulbs, leaves, bracts, pedicels, 0.5–0.8 cm flower buds, 2.0−2.5 cm flower buds, blooming flowers, sepals, petals, lips, and columns of C. goeringii. The RNA was used in de novo sequencing by Illumina HiSeq 2500. A 20 kb single-molecule real-time (SMRT) DNA library was constructed and sequenced on the PacBio Sequel platform. SMRTbell template preparation involved DNA concentration, damage repair, end repair, ligation of hairpin adapters, and template purification and was undertaken using AMPure PB Magnetic Beads (Pacific Biosciences). Young leaves of C. goeringii were used to construct the Hi-C sequencing library and sequenced on the MGISEQ-2000 platform. The plants were grown in Shaoxing, Zhejiang Province, China.
Genome assembly of C. goeringii and quality control
-
Genome assembly of C. goeringii was performed using Pacbio reads. First, Falcon[36] was used to correct the Pacbio raw reads, and then smartdenovo v1.0[37] was used to assemble the corrected reads. Due to the high error rate of the Pacbio reads, indel and SNP errors still existed in the assembly results. Illumina reads were used to correct the assembly results by pilon v1.22[38]. Genome size and heterozygosity were measured using jellyfish v2.1.4[39] and genomeScope[40] based on a 19-mer distribution. The total length of the assembly result was larger than the genome size estimated by k-mer analysis; trimDup was used to reduce the redundancy of the assembly results. SOAPnuke v2.1.0 was used to filter the Hi-C raw reads (parameter: filter -n 0.02 -l 20 -q 0.4 -G 2 -i -Q 2 --seqType 0) and obtain clean reads. The clean reads were mapped to the genome by Juicer[41], and the results were filtered to remove the misaligned reads. The genome sequence was preliminarily clustered, sequenced, and directed by 3d-dna[42]. The visualisation software Juicer box[41] was used to adjust, relocate, and cluster the genome sequence.The assembly quality and integrity of the genome were assessed by BUSCO v3[43].
Repeat, structural and functional annotation of genes
-
The repeat sequence annotation combined homolog and de novo prediction. In the homology-based prediction method, RepeatMasker v4.0.7 and RepeatProteinMask v.4.0.7[44] with the RepBase v21.12 database[45] (
http://www.girinst.org/repbase ) were used to find the known repeat sequences. In the de novo prediction method, RepeatModeler v.1.0.3[44] (http://www.repeatmasker.org/RepeatModeler ), LTR_FINDER v.1.06[46] (http://www.girinst.org/repbase ), and PILER v.1.3.4[47] were used to construct a de novo repeat sequence database, and the repeat sequences were searched in the genome by RepeatMasker. In addition, tandem repeat sequences were found in the genome by Tandem Repeats Finder v4.09[48].Gene prediction and functional annotation were conducted by a combination of homology-based prediction, de novo prediction, and transcriptome-based prediction methods. The homology-based method was performed by comparing the protein sequences of known homologous species (Gastrodia elata, Phalaenopsis equestris Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, and Zea mays) with the genome sequences of C. goeringii. Gene structure was predicted by Genewise v2.4.1[49]. Five-thousand genes with integral structure were randomly selected from the homologous predicted genes and used to train the de novo prediction software. Augustus[50] and SNAP[51] were used to construct the de novo gene prediction model of C. goeringii. Finally, combined with the RNA-seq data, the genome was annotated with maker v2.31.8 software[52], and the genes overlapping with repetitive sequence elements were removed; a total of 30,876 genes were obtained.
The protein sequences were annotated using seven annotation databases, namely GO[53], KEGG[54], KOG[55], InterPro[56], SwissProt[57], Nr, and TrEMBL[58]. The noncoding rRNAs were identified by aligning the rRNA template sequences from the Rfam[58] database against the genome using the BLASTN algorithm at an E-value of 1e-5. tRNAs were predicted using tRNAscan-SE 1.3.1[59], and other ncRNAs (miRNA and snRNA) were predicted by Infernal software (
http://infernal.janelia.org ) against the Rfam database.Gene family, WGD event and phylogenomic analysis
-
Gene families were identified in 17 species (C. goeringii, D. catenatum, P. equestris, G. elata, Apostasia shenzhenica, Asparagus officinalis, Brachypodium distachyo, O. sativa, S. bicolor, Ananas comosus, Phoenix dactylifera, Musa acuminata, Spirodela polyrhiza, A. thaliana, Populus trichocarpa, Vitis vinifera, and Amborella trichopoda) genomes by OrthoMCL v2.0.9[60]. A total of 266 single-copy gene families were identified. Single-copy genes shorter than 100 bp were removed, and a total of 160 genes were obtained to construct a supergene for phylogenetic relationships and divergence time analysis. The protein sequences were aligned by MUSCLE v3.8.31[61] and filtered by trimal[62]. The dataset was used to construct the phylogenetic tree by Ra×ML[63] with the nuclear acid substitution model GTRGAMMA.
The divergence time was conducted by MCMCTree in PAML 4.9[64] with the GTR model. The calibration time was selected as follows: O. sativa–B. distachyo (40–54 Mya)[65]; A. thaliana–P. trichocarpa (100–120 Mya)[66]; lower limit of divergence time of monocotyledons and dicotyledons (140 Mya)[67]; and upper limit of angiosperm formation time (200 Mya)[68]. The gene family expansion and contraction of 17 species were analysed by CAFE 4[69].
Genes in the collinear fragments are conserved in function and sequence; these genes also remain highly conserved during evolution. The protein sequences of C. goeringii and P. equestris were analysed to obtain the gene pairs in the collinear region using the default parameters of JCVI v0.9.14[70]. The Ks (synonymous substitutions per synonymous site) distribution analysis was used to estimate WGD events in the C. goeringii genome. DIAMOND[71] was used to conduct self-alignment on the protein sequences of C. goeringii with A. shenzhenica, D. catenatum, G. elata, P. equestris, A. officinalis, and C. goeringii and to extract the mutual optimal alignment in the alignment results. Codeml in the PAML package was used to calculate the Ks value[72].
Gene identification and expression
-
The MADS-box/MYB gene protein sequences of A. thaliana were downloaded from the TAIR database (
https://www.arabidopsis.org ). Then, a Blast search was performed against all protein sequences of C. goeringii in TBtools[73], and those with E-values less than 1e-5 were selected as the candidate proteins. The candidate proteins were submitted to NCBI BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi ) to filter the non-MADS-box/MYB proteins. The HMMER suite (http://hmmer.janelia.org ) was used to align the TPS protein sequences of C. goeringii against the hidden Markov model of the Pfam profiles of PF01397 and PF03936 (E value < 10−5)[74,75]. Further domain analysis was performed in NCBI CDD (Conserved Domain Database,http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi )[76] to confirm the presence and completeness of the candidate proteins. Multiple sequence alignment was performed in MEGA5.0[77], and a phylogenetic tree was constructed on the CIPRES website (https://www.phylo.org/portal2 ). The gene expression levels were indicated by FPKM on the transcriptome data. Resistance genes of C. goeringii were identified by HMMER V3.0 against the hidden Markov model of the NB-ARC domain (Pfam accession PF00931). The TIR and LRR domains were detected using the Pfam_Scan (−E 0.01 –domE 0.01). MARCOIL and paircoil were utilized for identification of the CC motif. Hps70 genes were identified by HMMER V3.0 against the hidden Markov model of the Hps70 domain (Pfam accession PF00012), and combined with the results of Blast to get the final gene set.Data availability
-
Genome sequences and whole-genome assemblies have been submitted to the National Center for Biotechnology Information (NCBI) database with BioProject accession number PRJNA749652.
This research was jointly funded by the project of Jiangsu agricultural science and technology innovation fund: 'Research on the breeding technique of new varieties with red flowers of spring orchids' (No. CX(19)3118); Research Fund of Agricultural Science Institute in Lixiahe area of Jiangsu Province (No. SJ(21)302); Platform for the protection and utilization of agricultural germplasm resources in Jiangsu Province: 'The germplasm resources nursery of Chinese orchids' (No. JSGB2018-01).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Ye Sun, Gui-Zhen Chen
- Supplemental Fig. S1 Genome size and heterozygosity estimation using 19 K-mer distribution.
- Supplemental Fig. S2 Intensity signal heat map of Hi-C chromosome.
- Supplemental Fig. S3 The sequence divergence rate of four different TEs using RepeatMasker annotation.
- Supplemental Fig. S4 The sequence divergence rate of four different TEs using de novo annotation.
- Supplemental Fig. S5 GO analysis enrichment of unique gene families in the C. goeringii genome.
- Supplemental Fig. S6 KEGG pathway enrichment of unique gene families in the C. goeringii genome.
- Supplemental Fig. S7 KOG pathway enrichment of unique gene families in the C. goeringii genome.
- Supplemental Fig. S8 The Venn diagram shows the annotation genes in Nr, InterPro, KEGG, SwissProt and KOG.
- Supplemental Fig. S9 Phylogenetic tree of 17 species. Phylogenetic tree was constructed using phase 1 loci of orthologous genes, with each branch length representing the evolutionary rate.
- Supplemental Fig. S10 Gene pairs in the collinear region among P. equestris and C. goeringii. A. Gene pairs in the collinear region of C. goeringii and C. goeringii. B. Gene pairs in the collinear region of C. goeringii and P. equestris.
- Supplemental Fig. S11 Phylogenetic tree of MADS genes of A. shenzhenica, A. thaliana and C. goeringii. The gene ID number begins with ‘AT’ to represent the gene ID of A. thaliana; the gene ID number begins with ‘Ashe’ to represent the gene ID of A. shenzhenica; the gene ID number begins with ‘Phe’ to represent the gene ID of P. equestris; the gene ID number begins with ‘Osa’ to represent the gene ID of O. sativa, and the gene ID number begins with ‘GL’ to represent the gene ID of C. goeringii.
- Supplemental Fig. S12 Expression of MADS-box genes in different tissues of C. goeringii. RT, root; LE, leaf; ST, stem; FL, flower; SP, sepals; PT, petals; LIP, lip; COL, column.
- Supplemental Fig. S13 Expression of MADS-box genes in different mutants of C. goeringii. NOS, sepals of normal flowers; NOP, petals of normal flowers; NOL, lip of normal flowers; NOC, column of normal flowers; CPS, sepals of column-like perianth mutant; CPP, petals of column-like perianth mutant; CPL, lip of column-like perianth mutant; CPC, column of column-like perianth mutant; LSS, sepals of lip-like sepal mutant; LSP, petals of lip-like sepal mutant; LSL, lip of lip-like sepal mutant; LSC, column of wild lip-like sepal mutant.
- Supplemental Fig. S14 Expression of MADS-box genes in the tepal-like leaves and the normal leaves of C. goeringii. A. Tepal-like leaf and normal green leaf. B. Expression profile of MADS-box genes. TL, tepal-like leaf, NL, normal green leaf.
- Supplemental Fig. S15 Expression analysis of six R2-R3 MYB genes related to flower colour in C. goeringii. PYS, sepals of pale-yellow flowers with purple-red spots; PYP, petals of pale-yellow flowers with purple-red spots; PYL, lip of pale-yellow flowers with purple-red spots; GYS, sepals of pale-yellow flowers with purple-red spots; GYP, petals of pale-yellow flowers with purple-red spots; GYL, lip of pale-yellow flowers with purple-red spots; PRS, sepals of pale-yellow flowers with purple-red spots; PRP, petals of pale-yellow flowers with purple-red spots; PRL, lip of pale-yellow flowers with purple-red spots.
- Supplemental Fig. S16 Evolutionary analysis of the TPS genes in C. goeringii.
- Supplemental Fig. S17 Differentially expressed genes related to chlorophyll synthesis in different colour leaves of C. goeringii. Normal green leaf, tissue of normal green leaf; Yellow, yellow tissue of yellow-green leaf mutant type; Green, green tissue of yellow-green leaf mutant type.
- Supplemental Table S1 K-mer statistics of genome sequencing results of C. goeringii.
- Supplemental Table S2 The statistics of PacBio sequencing data.
- Supplemental Table S3 Assembly statistics of the C. goeringii genome.
- Supplemental Table S4 BUSCO assessment of the C. goeringii genome.
- Supplemental Table S5 Hi-C sequence data statistics.
- Supplemental Table S6 The statistical results of Hi-C assembly.
- Supplemental Table S7 Chromosome length by Hi-C assembly.
- Supplemental Table S8 The statistical results of repeat sequences.
- Supplemental Table S9 Statistic of repeat sequence in C. goeringii.
- Supplemental Table S10 The prediction of gene structures of the C. goeringii.
- Supplemental Table S11 The number of protein-coding genes supported by de novo, transcriptome data and homology prediction.
- Supplemental Table S12 BUSCO assessment of annotation of C. goeringii genome.
- Supplemental Table S13 Statistics on the annotation of non-coding RNA of the C. goeringii genome.
- Supplemental Table S14 The statistical results of functional annotation.
- Supplemental Table S15 Statistical results of clustered gene families.
- Supplemental Table S16 GO enrichment of significantly expanded ( p < 0.05 ) gene families of the C. goeringii genome.
- Supplemental Table S17 KEGG pathway enrichment of significantly expanded ( p < 0.01) gene families of the C. goeringii genome.
- Supplemental Table S18 GO enrichment of unique gene families of the C. goeringii genome.
- Supplemental Table S19 KEGG enrichment of unique gene families of the C. goeringii genome.
- Supplemental Table S20 List of MADS-box genes identified in C. goeringii.
- Supplemental Table S21 List of resistant genes of three orchid species.
- Supplemental Table S22 List of Hsp70 genes of three orchid species.
- Supplemental Table S23 The enzymes related to floral colour, fragrance, and chlorophyll degradation regulatory networks in C. goeringii.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Sun Y, Chen G, Huang J, Liu D, Xue F, et al. 2021. The Cymbidium goeringii genome provides insight into organ development and adaptive evolution in orchids. Ornamental Plant Research 1: 10 doi: 10.48130/OPR-2021-0010 

The Cymbidium goeringii genome provides insight into organ development and adaptive evolution in orchids
- Received Date: 27 May 2021
- Accepted Date: 30 August 2021
- Published Online: 09 September 2021
Abstract: Cymbidium goeringii is one of the important ornamental orchids, but its high-quality genome has not been previously published. Here, we report a chromosome-level genome of C. goeringii and report the gene family expansion, and contraction of the C. goeringii genome and the regulation mechanism of MADS-box genes in floral organ development. We constructed the pathways of carotenoids and anthocyanins that contribute to the different flower colors of C. goeringii and the metabolic pathways of the main components of flower fragrance. Moreover, we found the genes that regulate colourful leaves and analyzed the resistance genes involved in the adaptive evolution of C. goeringii. Our results provide valuable genomic resources for the improvement of orchids and other ornamental plants.
-
Key words:
- Cymbidium /
- genome evolution /
- organs development /
- adaptive evolution /
- flower colour /
- floral scent