 Zhuo-Yu Cai1,2,3
Zhuo-Yu Cai1,2,3 Zheng-Yang Niu1,2,3
Zheng-Yang Niu1,2,3 You-Yuan Zhang1,4Yi-Hua Tong1,3,5
You-Yuan Zhang1,4Yi-Hua Tong1,3,5 Tien Chinh Vu6,7Wei Lim Goh8
Tien Chinh Vu6,7Wei Lim Goh8 Sarawood Sungkaew9Atchara Teerawatananon10
Sarawood Sungkaew9Atchara Teerawatananon10 Nian-He Xia1,3*
Nian-He Xia1,3*- 1Key Laboratory of Plant Resources, Conservation and Sustainable Utilization/Guangdong Provincial Key Laboratory of Digital Botanical Garden, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 3South China National Botanical Garden, Guangzhou, China
- 4Quality Management Office, Guiyang Vocational and Technical College, Guiyang, China
- 5State Key Laboratory of Plant Diversity and Specialty Crops, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
- 6Vietnam National Museum of Nature, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 7Graduate University of Science and Technology, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 8Faculty of Science, Universiti Tunku Abdul Rahman, Jalan Universiti, Kampar, Perak, Malaysia
- 9Department of Forest Biology, Faculty of Forestry, Kasetsart University, Bangkok, Thailand
- 10Natural History Museum of Thailand, National Science Museum, Pathum Thani, Thailand
Neomicrocalamus and Temochloa are closely related to bamboo genera. However, when considered with newly discovered and morphologically similar material from China and Vietnam, the phylogenetic relationship among these three groups was ambiguous in the analyses based on DNA regions. Here, as a means of investigating the relationships among the three bamboo groups and exploring potential sources of genomic conflicts, we present a phylogenomic examination based on the whole plastome, single-nucleotide polymorphism (SNP), and single-copy nuclear (SCN) gene datasets. Three different phylogenetic hypotheses were found. The inconsistency is attributed to the combination of incomplete lineage sorting and introgression. The origin of newly discovered bamboos is from introgressive hybridization between Temochloa liliana (which contributed 80.7% of the genome) and Neomicrocalamus prainii (19.3%), indicating that the newly discovered bamboos are closer to T. liliana in genetics. The more similar morphology and closer distribution elevation also imply a closer relationship between Temochloa and newly discovered bamboos.
1 Introduction
The bamboo tribe Bambuseae (Poaceae: Bambusoideae), also known as the paleotropical and neotropical woody bamboos, has a significant role in ecology and socio-economy (Kambale et al., 2022) and is considered a phylogenetically and taxonomically challenging group (Goh et al., 2013; Liu et al., 2020). The cytonuclear discordance is a reflection of reticulate evolution, which makes the paleotropical woody bamboos a phylogenetically recalcitrant group (Liu et al., 2020). The discordance could arise from evolutionary processes, such as hybridization, introgression, and incomplete lineage sorting (ILS) (Smith et al., 2015; Stull et al., 2020). In previous studies, it has been demonstrated that assessing gene flows (hybridization and introgression) and ILS could help us reach a better understanding of closely related taxa (García et al., 2017; Morales-Briones et al., 2018). For paleotropical woody bamboos, some hybrids have been reported among erect bamboos (Goh et al., 2011; Goh et al., 2018); however, hybridization/introgression and ILS, which are presumed as underlying causes for their complex evolutionary history (Goh et al., 2013), have been poorly explored in climbing bamboos.
Neomicrocalamus Keng f. is a small genus of climbing or scrambling bamboos, comprising three species and distributed in Bhutan, southwest China, northeast India, and Vietnam (Li and Stapleton, 2006; BPG [Bamboo Phylogeny Group], 2012; Vorontsova et al., 2016). Neomicrocalamus dongvanensis T.Q. Nguyen from Vietnam may not belong to this genus as it possesses erect culms (Nguyen, 1991; Stapleton et al., 1997). This genus has often been regarded as related to Racemobambos Holttum, another group of climbing or scrambling bamboos distributed in Malesia (Holttum, 1956; Wen, 1986; Stapleton, 1994). However, in the phylogenetic analyses inferred with plastid and nuclear fragments, Neomicrocalamus has been shown to be distantly related to Racemobambos and sister to Temochloa S. Dransf. (Ruiz-Sanchez and Sosa, 2015; Zhou M.Y. et al., 2017).
Temochloa is a monotypic genus in Thailand and possibly Laos (Dransfield, 2000; Zhou M.Y. et al., 2017). Up to now, little is known about it. Some crucial morphological characters are still unavailable, such as lodicules, stamens, and fruits. As such, it was not possible to place Temochloa in a subtribal classification of Southeast Asian woody bamboos (Wong et al., 2016). Likewise, it was regarded as having an affinity to Racemobambos (Dransfield, 2000). However, Temochloa possesses some characteristics in common with Neomicrocalamus in morphology and biogeography. Both of them are climbing bamboos in limestone areas and have short-necked pachymorph rhizomes and branch complements with many short and subequal branches with an occasional dominant central branch that reiterates and approaches the size of the culm. In phylogenetic analyses based on plastid regions, it was recovered close to the Bambusa–Dendrocalamus–Gigantochloa (BDG) complex and sister to Neomicrocalamus (Sungkaew et al., 2009; Kelchner and BPG [Bamboo Phylogeny Group], 2013; Zhou M.Y. et al., 2017). However, substantial conflicting signals were detected in the organelle DNA dataset (Zhou M.Y. et al., 2017).
During fieldwork in North Vietnam, some climbing bamboos came to our attention, but no flowering material was obtained until subsequent exploration in a neighboring area in China (Table S1). The Chinese material (collection number: BH85) is significant among these newly discovered bamboos as it is the only one containing flowers and fruits. After consulting the literature, potential placements in Neomicrocalamus or Temochloa were considered for these newly discovered bamboos. These three groups share a number of similar morphological characters, prompting an investigation of their phylogenetic relationships in order to better understand if taxonomic placements may be better justified or improved.
However, the phylogeny based on DNA fragments shows an ambiguous relationship among these three groups. The use of single-nucleotide polymorphism (SNP) datasets has been demonstrated to give a better resolution in bamboo phylogeny (Liu et al., 2020; Guo et al., 2021; Liu et al., 2022). In addition, the multispecies coalescent model used to reconstruct species trees with large numbers of nuclear genes has also much-improved accuracy of phylogenetic inference (McCormack et al., 2009; Smith et al., 2015; Liu et al., 2021). In this study, we aim to 1) examine the relationship among these two genera and the newly discovered bamboos based on the whole plastome, SNP, and single-copy nuclear (SCN) gene datasets with both concatenation and coalescence methods, and 2) assess the potential sources of plastid/nuclear discordance and the gene tree/species tree incongruence.
2 Materials and methods
2.1 Taxon sampling
For inference of the systematic position, phylogenetic reconstruction was based on plastid loci referred to in the analyses of Zhou M.Y. et al., (2017) and Haevermans et al. (2020). Finally, 23 samples were involved in the reconstruction, among which 15 samples, representing 14 species (including one variety) from nine genera, were downloaded from GenBank. Eight samples were newly sequenced in this study. Guadua angustifolia Kunth was set as the outgroup for this analysis.
The eight newly sequenced samples were also used to conduct further analyses on the basis of the complete plastome, SNP, and SCN gene datasets. For the plastome, SNP, and SCN phylogenetic tree reconstruction, another sample was added as the outgroup, Bonia levigata (L.C. Chia, H.L. Fung & Y.L. Yang) N.H. Xia. All sample details are provided in Tables S1 and S2.
2.2 DNA extraction and sequencing
Total genomic DNA was isolated from silica-dried healthy leaves using the TIANGEN Genomic DNA Extraction Kit (TIANGEN, Beijing, China). DNA samples of concentration meeting the standard (≥1 μg) were randomly sheared into fragments using Covaris M220 (Covaris, Woburn, MA, USA). Fragments of 350 bp were enriched using PCR, and the paired-end (2 × 150 bp) reads were generated in Novogene (Beijing, China) using the NovaSeq 6000 platform. As a result, approximately 20 Gb genome skimming data were generated for each sample. Among species with already published bamboo genome data, Bonia amplexicaulis (L.C. Chia, H.L. Fung & Y.L. Yang) N.H. Xia, with a whole genome size of 0.848 Gb (Guo Z.H. et al., 2019), is the species closest to the taxa included in the present investigation (Zhou M.Y. et al., 2017). Hence, the expected sequencing coverage for our samples would be approximately 23.58× (20 Gb/0.848 Gb). For confirmation of the actual coverage, Jellyfish 2.2.3 (Marçais and Kingsford, 2011) and GenomeScope 2.0 (Ranallo-Benavidez et al., 2020) were used to estimate genome size for each newly sequenced sample (including the outgroup).
2.3 Plastome assembly, annotation, and plastid locus extraction
All DNA regions were extracted from plastomes using a python script “get_annotated_regions_from_gb.py” (https://github.com/Kinggerm/PersonalUtilities/). Eventually, following the procedures of Zhou M.Y. et al. (2017), 18 plastid loci were selected (rpl32-trnL, trnT-trnL, trnL-trnF, psbA-trnH, rpl16 intron, rps16-trnQ, trnC-rpoB, trnD-trnT, rps16 intron, ndhF [3′ end], matK, atpB-rbcL, psbM-petN, trnS-trnfM, ycf4-cemA, trnG-trnT, rps15-ndhF, and rbcL-psaI). All plastid loci were concatenated for phylogenetic analysis.
The filtered clean reads were used for de novo assembly of complete plastomes using the GetOrganelle v.1.6.2 pipeline (Jin et al., 2020) with six k-mer values: 21, 45, 65, 85, 105, and 125. Subsequently, the filtered plastid reads were transferred to Bandage v.0.8.1 (Wick et al., 2015) for the visualization process. Two opposite plastid sequences exported from Bandage were aligned with the reference sequence Bonia saxatilis (L.C. Chia, H.L. Fung & Y.L. Yang) N.H. Xia (GenBank accession No. MK679779), and the one matching the reference was annotated using PGA (Qu et al., 2019). Finally, plastomes were manually corrected in Geneious v.9.1.4 (Kearse et al., 2012).
2.4 SNP dataset construction
The latest high-quality genome sequence of Dendrocalamus latiflorus Munro (Zheng et al., 2022) was selected as the chromosome-level reference to build an index using SAMtools v.1.9 (Danecek et al., 2021) and Picard v.2.27.3 (Broad, 2019). After the filtration of low-quality data, clean reads were processed with the removal of duplicates using Fastuniq v.1.1 (Xu et al., 2012). Newly filtered paired reads were aligned to the reference using Bowtie2 v.2.4.4 (Langmead and Salzberg, 2012) with the minimum acceptable alignment score set as L, 0.3, 0.3. SAMtools was invoked to sort out alignments in binary alignment format (BAM) files. Picard was used to remove duplicates again with the function “MarkDuplicates”. GATK v.4.2.2.0 (van der Auwera and O'Connor, 2020) was used to anchor variable sites including SNP and InDel in the genomic variant call format (GVCF) file by the joint-calling method “HaplotypeCaller” with the minimum assembly region size as 10 and k-mer from 10 to 25. After completion of variant calling, the tool “CombineGVCFs” in GATK was performed to combine all the GVCF files. The tool “GenotypeGVCFs” was then used to identify all the joint-called variants. After that, filtration of low-quality SNPs was conducted with the tool “VariantFiltration” with the following parameters: QD < 2.0, MQ < 40.0, FS > 60.0, SOR > 3.0, MQRankSum < −12.5, and ReadPosRankSum < −8.0. Then, the tool “SelectVariants” was run to extract raw SNPs. After extraction, plink v.1.90b4.6 (Purcell et al., 2007) was used to filter low-quality SNPs with the parameter “geno” set as 0.1 and “maf” set as 0.01. Filtered variants were then pruned with the parameter “indep-pairwise” set as 50, 10, and 0.2, representing its window size, a variant count to shift the window, and pairwise r2 threshold for SNPs, respectively. Finally, a clean SNP dataset was generated, and the GVCF file was transferred to a PHYLIP file for phylogenetic analysis using the python script “vcf2phylip.py” (Ortiz, 2019).
2.5 SCN dataset construction
The protein-coding sequences of five previously published bamboo genomes, namely, Phyllostachys edulis (Carrière) J. Houz. (Zhao et al., 2018), B. amplexicaulis, G. angustifolia Kunth, Olyra latifolia L., and Raddia guianensis (Brongn.) Hitchc. (Guo Z.H. et al., 2019), were used to identify conserved orthologous genes based on the Liliopsida and Poales datasets of BUSCO v.4.1.3 (Manni et al., 2021). Then, the selected orthologous genes of these five bamboo genomes were used to further identify the single-copy nuclear genes using OrthoFinder v 2.4.0 (Emms and Kelly, 2019). A total of 443 genes were generated and set as the reference gene dataset. The filtered reads were mapped to the reference and assembled using the software HybPiper v 1.3 (Johnson et al., 2016) with default parameters. Finally, following the previous study (Zhou et al., 2022), these assembled genes, which are longer than 200 bp, were used in the subsequent analyses.
2.6 Phylogenetic analysis
Each dataset (plastid locus, whole plastome, SNP, single SCN gene, and concatenated SCN genes) was aligned using MAFFT v.7.450 (Katoh and Standley, 2013). For each data matrix, both maximum likelihood (ML) and Bayesian inference (BI) analyses were conducted. Maximum likelihood analysis was carried out in RAxML-HPC v.8.2.10 (Stamatakis, 2014), with a GTRGAMMA nucleotide substitution model and 1,000 rapid bootstrap replicates. Bayesian inference analysis was carried out using MrBayes v.3.2.7 (Ronquist et al., 2012). The HKY+I+G model, deduced using MrModeltest2 v.2.4 (Nylander, 2004) under the Akaike information criterion (AIC) (Posada and Buckley, 2004), was selected for the plastid locus matrix, as well as the plastome matrix, the GTR model was selected for the SNP matrix, and the GTR+I+G model was selected for the SCN gene concatenated matrix. At least 30,000,000 generations were run to ensure that the average standard deviation of split frequencies was lower than 0.01, with the sampling frequency set as 100 generations. The first 25% of sampled trees were discarded as burn-in.
To infer species trees, two different approaches were used. The first method was conducted using ASTRAL-III v.5.7.8 (Zhang et al., 2018). Individual SCN gene trees were estimated using RAxML-HPC v.8.2.10 (Stamatakis, 2014) with a GTRGAMMA model and 500 rapid bootstrap replicates. Branches of each gene tree with support lower than 50% were removed to improve the accuracy of species tree inference. Then, individual gene trees and their bootstrap replicates were used to estimate species trees with 500 coalescent bootstrap replicates. SVDquartets v.1.0 (Chifman and Kubatko, 2014) implemented in PAUP v.4.0a169 (Swofford, 2003) was the second method that utilizes the concatenated SCN gene matrix to infer the species tree. The clade support was assessed using 1,000 bootstrap replicates.
The final phylogenetic results were visualized using FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
2.7 Network analysis
In order to investigate potential conflicting signals within the plastid locus matrix, the Neighbor-Net algorithm based on uncorrected P-distances was performed with SplitsTree4 v.4.18.1 (Huson and Bryant, 2006).
2.8 Coalescent simulation
To test if the discordance between plastome tree and nuclear trees could be explained by ILS alone, we conducted coalescent simulations following previous studies (García et al., 2017; Morales-Briones et al., 2018; Zhou et al., 2022). The ASTRAL-III tree was chosen as a guide tree for the gene tree simulation using DENDROPY v3.12.1 (Sukumaran and Holder, 2010). To simulate plastid gene trees, branch lengths of the ASTRAL-III tree were scaled by 12 to account for organellar inheritance as paleotropical woody bamboos are hexaploidy, and the effective population size of the plastome is generally expected to be one-twelfth that of the nuclear genome given the assumptions of equal sex ratios, haploidy (homoplasmic), and uniparental inheritance (McCauley, 1994; Stull et al., 2020). Finally, 1,000 gene trees were simulated under the coalescent model. The clade frequencies of simulated trees were summarized on the plastome tree inferred using RAxML with PHYPARTS (Smith et al., 2015). In a scenario of ILS alone, the clades in the empirical plastome tree should be present in the simulated trees with high frequency. If hybridization exists, the clades of the empirical plastome trees should be absent or at very low frequency in the simulated trees (García et al., 2017; Morales-Briones et al., 2018; Zhou et al., 2022).
2.9 Gene flow analyses
To detect potential gene flows between lineages, the D-statistic test was performed in Dsuite v.0.5 (Malinsky et al., 2021). Each lineage was represented by all samples of each clade. B. levigata was treated as the outgroup. Z-scores greater than three are generally interpreted as strong evidence of gene flow; otherwise, ILS cannot be excluded as an explanation for ancient polymorphisms at maximum probability (Eaton and Ree, 2013; Ye et al., 2021).
To further explore the reticulate evolutionary history, SnaQ (Solís-Lemus and Ané, 2016) implemented in PhyloNetworks v.0.12.0 (Solís-Lemus et al., 2017) was used to infer species networks. The input was SCN gene trees estimated using RAxML. Analyses allowing for 0–4 hybridization (h) events were performed using 10 independent runs. The best network was selected when pseudolikelihood scores reached a nearly constant level.
3 Results
3.1 Basic features of three datasets
For the newly sequenced samples, the estimated genome size and the coverage ranged from 747.32 Mb to 927.72 Mb and from 21.56× to 26.76×, respectively (Table S3).
The dataset for the combined and aligned matrix of 18 plastid loci across 23 samples consisted of 18,484 bp. A total of 787 (4.26%) variable sites were found, of which 301 (1.63%) were parsimony informative sites and 486 (2.63%) were singleton variable sites. Missing data accounted for 5.5% of the entire matrix.
The plastome size ranged from 138,292 bp to 139,510 bp. The alignment of the plastome of nine samples comprised 140,288 bp, characterized using only 976 (0.69%) variable sites, including 327 (0.23%) parsimony informative sites and 649 (0.46%) singleton variable sites. Missing data accounted for only 0.7% of the entire matrix.
For the SNP data, the matrix was 1,651 bp in total length with 1,490 (90.25%) variable sites, including 279 (16.90%) parsimony informative sites and 1,211 (73.35%) singleton variable sites. The SNP data matrix did not have missing data.
A total of 358 SCN genes were obtained for each sample. The concatenated matrix for 358 SCN genes consisted of 618,938 bp, featuring 48,971 (7.91%) variable sites, of which 21,237 (3.43%) were parsimony informative sites and 27,734 (4.48%) were singleton variable sites. For the whole matrix, the missing data were 5.3%.
3.2 Systematic position inferred using 18 plastid loci
Inference of phylogenetic relationships using plastid loci conducted with different methods (ML and BI) yielded identical topologies but different support values for some nodes (Figure 1). Newly sequenced Neomicrocalamus samples clustered with published Neomicrocalamus samples (Figure 1), forming a well-supported Neomicrocalamus clade (Bayesian inference posterior probability [PP]/bootstrap [BS], 1/100). The two Temochloa liliana S. Dransf. samples were recovered together with PP/BS 1/100 support and sister to Temochloa sp. (PP/BS 1/66), together making up the Temochloa clade. The three newly discovered bamboo samples formed a clade with PP/BS 1/100 support and, together with Neomicrocalamus spp. and Temochloa spp., represent a monophyletic lineage with high support (PP/BS 1/93). However, the relationship among the three clades of this lineage was unresolved as one of the pivotal nodes was not fully supported (PP/BS 0.984/49).
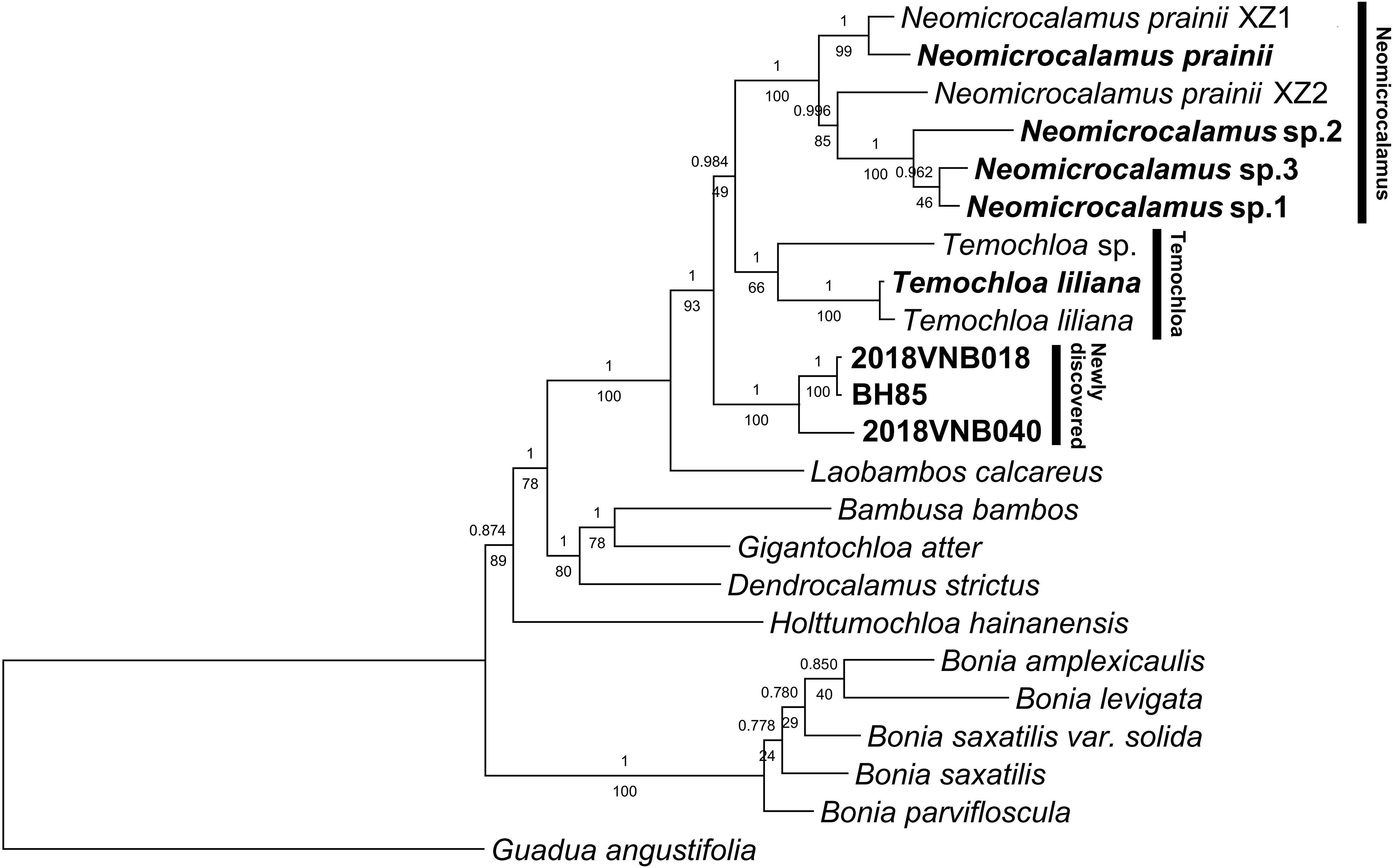
Figure 1 Majority rule (50%) consensus phylogenetic tree based on a combined plastid dataset of 18 loci. Both Bayesian inference (BI) and maximum likelihood (ML) analyses were conducted. Numbers above and below branches indicate Bayesian inference posterior probability (PP) and bootstrap (BS), respectively. BH85, 2018VNB018, and 2018VNB040 represent the newly discovered bamboos; these and other accessions freshly obtained for this analysis are indicated in bold.
The network analysis revealed a splitting pattern similar to the phylogenetic tree topology, in which Neomicrocalamus, Temochloa, and the newly discovered bamboo clusters were connected by many parallel short edges, indicating character conflicts among these three clades (Figure S1).
3.3 Relationship revealed with plastome, SNP, and SCN
As with the plastid locus dataset, ML and BI analysis methods produced identical topologies. However, three backbone topologies were revealed with different datasets and methods: Topology A (recovered using plastome), Topology B (SNP and ASTRAL-III), and Topology C (concatenated SCN and SVDquartets).
In the well-resolved plastome phylogeny, with almost every node being fully supported (PP/BS 1/100), the newly discovered bamboo clade was sister to the Neomicrocalamus clade, and the Temochloa clade was in turn sister to both of them (Figure 2A). The SNP phylogeny was also well-resolved except for the relationship within the Neomicrocalamus clade. The clade comprising the newly discovered bamboos aligned with the Temochloa clade as a monophyletic lineage with PP/BS 0.955/89 support, which was sister to the Neomicrocalamus clade (Figure 2B). In the phylogenetic tree inferred with the concatenated SCN genes, the Temochloa clade and the Neomicrocalamus clade together form a well-supported (PP/BS 1/99) monophyletic lineage. The newly discovered bamboo clade is sister to them (Figure 2C).
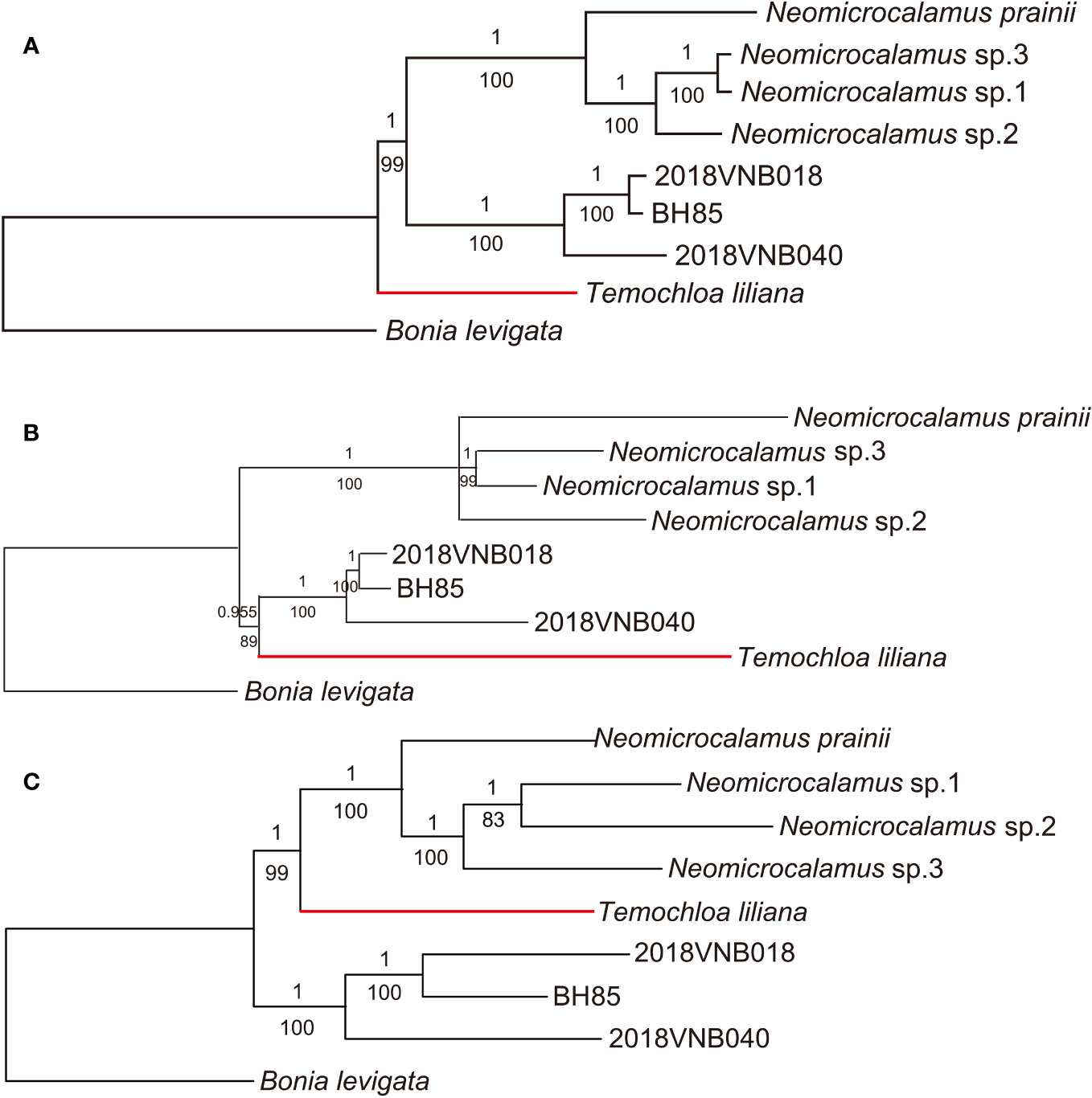
Figure 2 Phylogenetic trees of three groups of climbing bamboos based on the whole plastome (A), the single-nucleotide polymorphism (SNP) (B), and the concatenated single-copy nuclear (SCN) genes (C). Both Bayesian inference (BI) and maximum likelihood (ML) analyses were conducted for each dataset. Numbers above and below branches indicate Bayesian inference posterior probability (PP) and bootstrap (BS), respectively. BH85, 2018VNB018, and 2018VNB040 represent the newly discovered bamboos. The red branches indicate the position of Temochloa.
In the species trees, the backbone topology recovered using SVDquartets is the same as the concatenated SCN gene tree. However, the support value for the sister group relationship of the Temochloa clade and the Neomicrocalamus clade is not high enough (BS 74 < 75) (Figure 3). However, the ASTRAL-III found the Temochloa clade is sister to the newly discovered bamboo clade with good support (BS 92), in agreement with the SNP phylogeny.
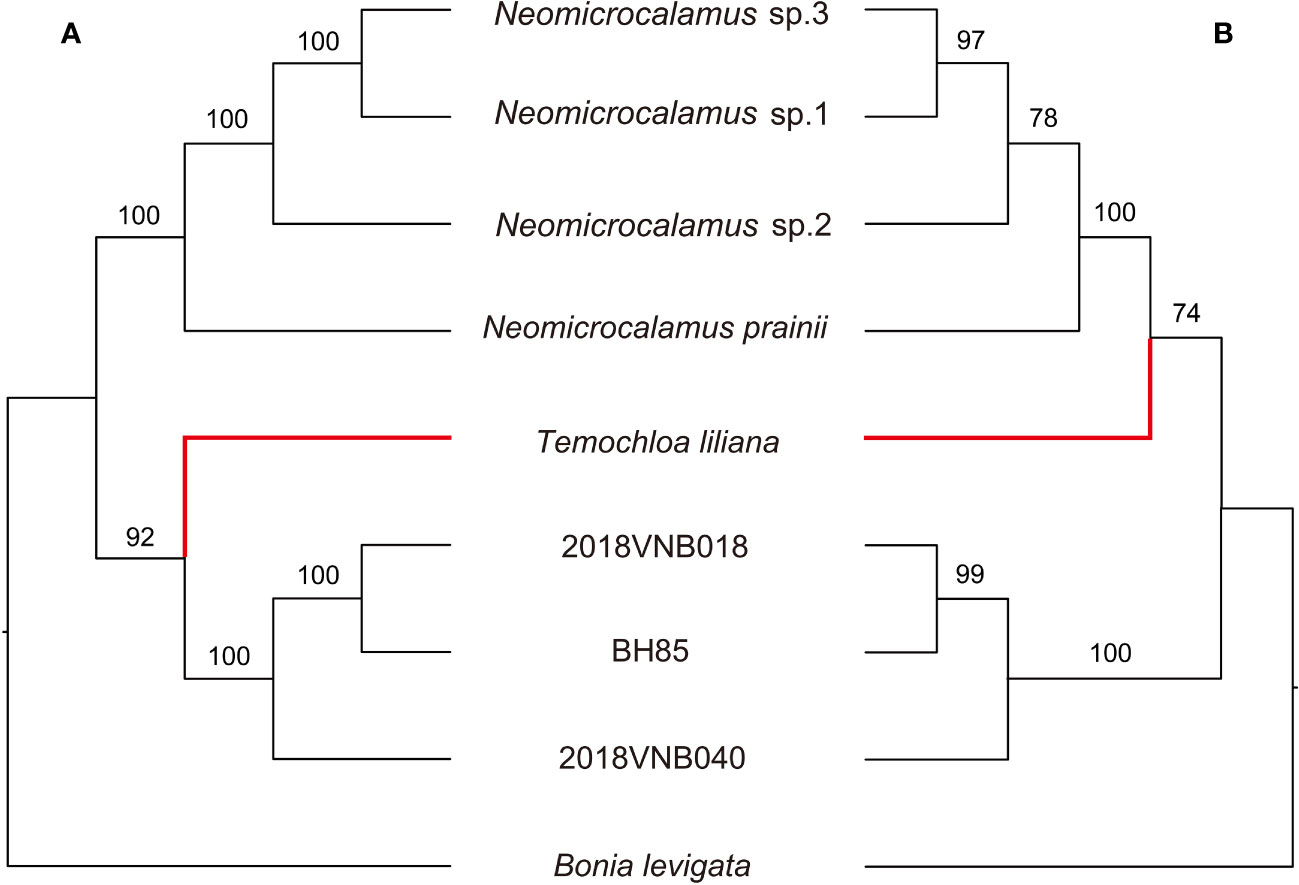
Figure 3 Tanglegram comparing the species trees inferred in ASTRAL-III (A) and SVDquartets (B). Numbers above branches indicate bootstrap (BS). BH85, 2018VNB018, and 2018VNB040 represent the newly discovered bamboos. The red branches indicate the position of Temochloa.
Furthermore, the relationship within the newly discovered bamboo clade is always identical among the five phylogenies. Although Neomicrocalamus prainii (Gamble) Keng f. is always sister to the other three Neomicrocalamus species, the position of Neomicrocalamus sp.2 is varied. In most of the phylogenies, Neomicrocalamus sp.1 is sister to Neomicrocalamus sp.3; however, in the concatenated SCN gene phylogeny, the position of Neomicrocalamus sp.3 is replaced by Neomicrocalamus sp.2.
3.4 Assessment ILS and gene flow
The plastid gene trees produced by the coalescent simulations resembled the empirical plastome tree. After summarizing simulated plastid gene trees onto the empirical plastome tree, most clade frequencies were at or near 100%, except for the clade of sister groups, the Neomicrocalamus group, and the newly discovered bamboo group, which is only 15.5% (Figure 4).
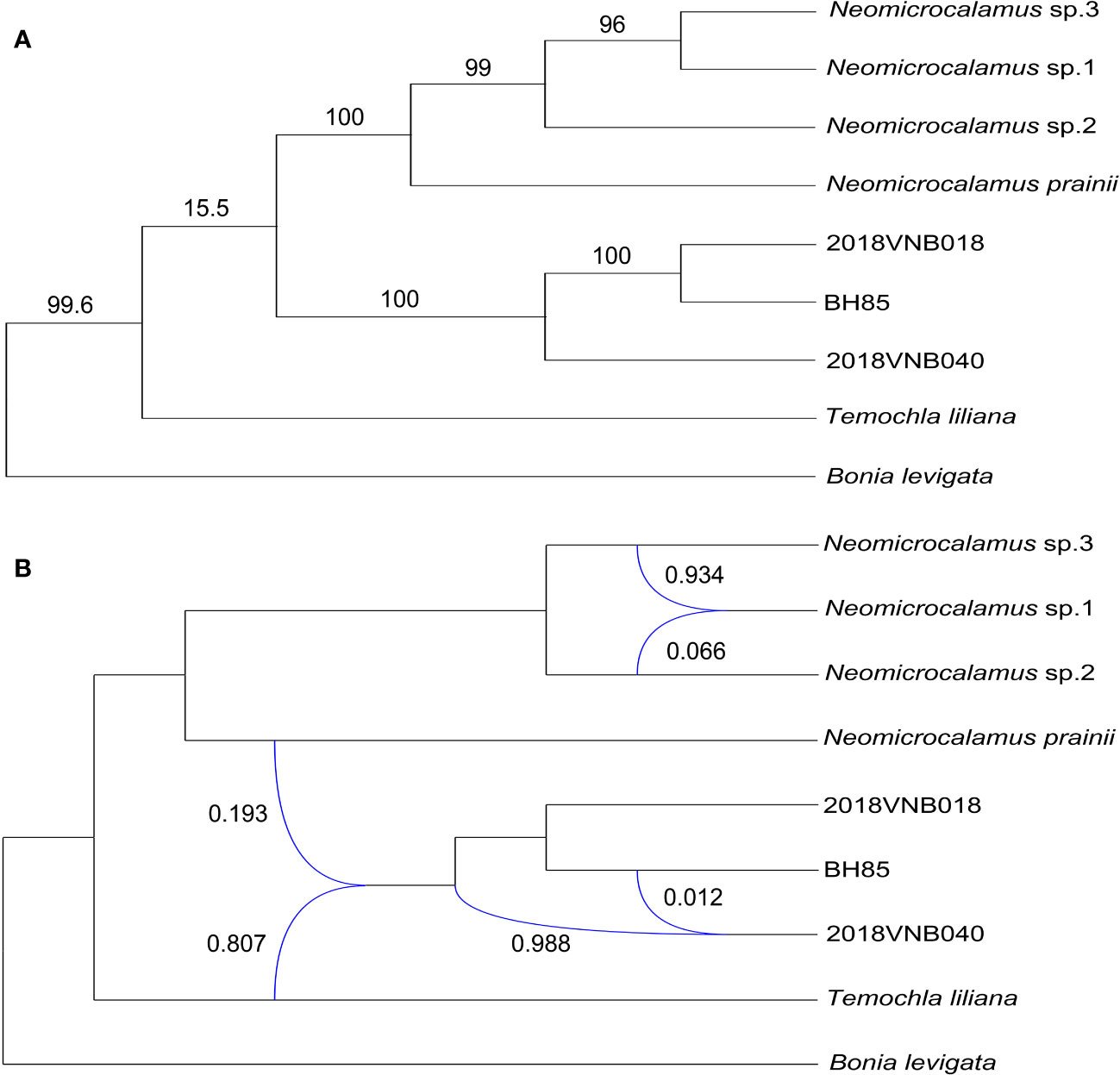
Figure 4 (A) Plastome phylogeny inferred with maximum likelihood; numbers above branches represent clade frequencies of the simulated gene trees. (B) Inference of gene flows; blue lines indicate hybrid edges, and numbers near blue lines are vectors of inheritance probabilities (γ).
From the D-statistic test, the Z-score was 1.63 lower than 3, which indicates both gene flows and ILS could be because of ancient polymorphisms (Table S4). However, the p-value (0.101 > 0.05) suggests that the result is only for reference.
Finally, three hybridization/introgression events were inferred (Figure 4 and Table S5). The results show that the newly discovered bamboo group inherited 80.7% of its genome from T. liliana and inherited 19.3% from N. prainii (Figure 4). The other two events are located within the Neomicrocalamus group and the newly discovered bamboo group, respectively. Both of them show largely unequal (ca. 95% vs. ca. 5%) gene flows between parents.
4 Discussion
In the previous phylogenetic analyses, Neomicrocalamus and Temochloa were revealed as closely related genera (sister groups) (Zhou M.Y. et al., 2017), which is also supported by some morphological and biogeographic evidence, such as short-necked pachymorph rhizomes and climbing culms endemic to limestone areas. After adding more accessions into the matrix of 18 plastid loci, the relationships among the main groups were congruent with those found in a former study (Zhou M.Y. et al., 2017), and the close relationship of these two genera is further confirmed, but these two genera together with the third bamboo group formed a three-clade polytomy (Figure 1) accompanied with substantial conflicting signals (Figure S1).
In the further phylogenetic analyses, three backbone topologies were recovered: Topology A (recovered by plastome), Topology B (SNP and ASTRAL-III), and Topology C (concatenated SCN and SVDquartets). The differences among topologies show the plastid/nuclear discordance and the gene tree/species tree incongruence. For the incongruence between two species trees ASTRAL-III and SVDquartets, we view the ASTRAL-III topology as more likely to be accurate as in the SVDquartets topology, the sister relationship between Temochloa clade and Neomicrocalamus clade is uncertain (low BS).
The cytonuclear discordance has been widely reported in plants (Mugrabi De Kuppler et al., 2015; Scheunert and Heubl, 2017; Soltis et al., 2019), as well as in Bambusoideae (Zhang et al., 2012; Wang et al., 2017; Guo C. et al., 2019). Our results of the coalescent simulation suggest that ILS was at play in the speciation of these taxa in this study, causing the cytonuclear discordance (Stull et al., 2020). Furthermore, the D-statistic test results (Z-score value < 3) also could not reject the existence of ILS (Eaton and Ree, 2013). The rapid evolutionary radiation and the long generation time of woody bamboos (Janzen, 1976; Guo Z.H. et al., 2019) are expected to maintain much of their ancestral polymorphism implying opportunities for extensive ILS (Pease and Hahn, 2015; Zhou Y. et al., 2017). Moreover, the hybridization/introgression events identified in this case also could be one of the reasons for the phylogeny incongruence (Linder and Rieseberg, 2004; Folk et al., 2017). In view of the largely unequal proportion of genes contributed by each parental lineage inferred by PhyloNetworks, the gene flows detected here are better interpreted as introgression (Solís-Lemus et al., 2017). The gene flow analyses suggest that the newly discovered bamboos originated from the introgressive hybridization between T. liliana and N. prainii, with T. liliana contributing 80.7% of the genome. In other words, genetically, the newly discovered bamboos are closer to T. liliana.
The similarity between the newly discovered bamboos and Temochloa is also indicated by another two aspects. In the biogeography, the newly discovered bamboos share the same distribution elevation with Temochloa 50–250 m, rarely reaching 700 m, whereas the Neomicrocalamus taxa are hitherto only found above 1,000 m (Dransfield, 2000; Li and Stapleton, 2006). This somehow interprets why T. liliana contributed a much higher genome component to the newly discovered bamboos, as gene flow is expected to happen more commonly among neighboring populations than distantly located populations (Petit and Excoffier, 2009; Zhou Y. et al., 2017). Morphologically, the newly discovered bamboos and Temochloa have nearly circular primary branch buds and shallowly grooved culm leaf sheaths, which are distinguished from the lanceolate buds and plane culm leaf sheaths of Neomicrocalamus (Dransfield, 2000; Li and Stapleton, 2006). On balance, the newly discovered bamboos have a closer relationship with Temochloa.
The phylogenetic network would help visualize clearer evolutionary relationships when reticulation events are involved (Morales-Briones et al., 2018). Empirically, the bifurcating phylogenetic tree inferred with SNP is more consistent with the evidence from morphology conventionally used to form classifications and genus circumscriptions for bamboos (Liu et al., 2020; Guo et al., 2021; Liu et al., 2022). In view of the limitation of bifurcating phylogenetic trees, the monophyletic group comprising Temochloa and the newly discovered bamboos, recovered in the SNP tree and ASTRAL-III species tree, seems to be the best classification for these two groups; as aforementioned, they are more closely related. However, overall, these three groups are closely related phylogenetically and morphologically. Therefore, for a more comprehensive understanding of their relationships, more evidence from all aspects should be investigated, such as the stamen of Temochloa, which is still unknown (Dransfield, 2000).
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number: OQ791227 OQ791228 OQ791225 OQ791224 OQ791221 OQ791226 OQ791222 OQ791223 OQ791229 (more details in Table S1). And it is also included in the Supplementary Material.
Ethics statement
All plant species used in the present study are not specially protected. The collections were permitted by the National Nature Reserve of Yarlung Tsangpo Grand Canyon, China; the Ba Be National Park, Vietnam; and the Klong Phanom National Park, Thailand. All plant experiments were conducted in accordance with relevant institutional, national, and international guidelines and legislation.
Author contributions
NX: Conceptualization, Funding acquisition, Writing – review & editing. ZC: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. ZN: Formal analysis, Methodology, Software, Writing – review & editing. YZ: Investigation, Writing – review & editing. YT: Investigation, Writing – review & editing. TV: Investigation, Writing – review & editing. WG: Formal analysis, Writing – review & editing. SS: Investigation, Writing – review & editing. AT: Investigation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (Grant No. 32270227), the Regional International Cooperation Project of Southeast Asia Biodiversity Research Institute, Chinese Academy of Sciences (Grant No. 2016CASSEABRIQG008), and the China Scholarship Council (File No. 202104910377).
Acknowledgments
We are grateful to Si-Jin Zeng for his kind assistance in the phylogenetic analysis and Mr. Van Da Nguyen from the Vietnam National Museum of Nature and Dr. Shi-Jin Li and Hai-Shan Chen from South China National Botanical Garden for their assistance in the fieldwork. Khoon Meng Wong (Singapore Botanic Gardens) assisted with some aspects of English.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1274337/full#supplementary-material
References
Bamboo Phylogeny Group (2012). An updated tribal and subtribal classification of the bamboos (Poaceae: Bambusoideae). J. Ame.r Bamboo Soc 24 (1), 1–10.
Broad Institute (2019). Picard Toolkit (Broad Institute, GitHub repository). Available at: https://broadinstitute.github.io/picard/.
Chifman, J., Kubatko, L. (2014). Quartet inference from SNP data under the coalescent model. Bioinformatics 30 (23), 3317–3324. doi: 10.1093/bioinformatics/btu530
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Pollard, M. O., Whitwham, A., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, 1–4. doi: 10.1093/gigascience/giab008
Dransfield, S. (2000). Temochloa, a new bamboo genus (Poaceae-Bambusoideae) from Thailand. Thai For. Bull. (Botany) 28, 179–182.
Eaton, D. A. R., Ree, R. H. (2013). Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62 (5), 689–706. doi: 10.1093/sysbio/syt032
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi: 10.1186/s13059-019-1832-y
Folk, R. A., Mandel, J. R., Freudenstein, J. V. (2017). Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 66 (3), 320–337. doi: 10.1093/sysbio/syw083
García, N., Folk, R. A., Meerow, A. W., Chamala, S., Gitzendanner, M. A., de Oliveira, R. S., et al. (2017). Deep reticulation and incomplete lineage sorting obscure the diploid phylogeny of rain-lilies and allies (Amaryllidaceae tribe Hippeastreae). Mol. Phylogenet. Evol. 111, 231–247. doi: 10.1016/j.ympev.2017.04.003
Goh, W. L., Chandran, S., Franklin, D. C., Isagi, Y., Koshy, K. C., Sungkaew, S., et al. (2013). Multi-gene region phylogenetic analyses suggest reticulate evolution and a clade of Australian origin among paleotropical woody bamboos (Poaceae: Bambusoideae: Bambuseae). Plant Syst. Evol. 299 (1), 239–257. doi: 10.1007/s00606-012-0718-1
Goh, W. L., Chandran, S., Kamiya, K., Wong, K. M. (2011). A natural hybrid between Dendrocalamus pendulus and Gigantochloa scortechinii (Poaceae: Bambusoideae: Bambuseae) in Peninsular Malaysia. Gard. Bull. Singapore 62 (2), 11–26.
Goh, W. L., Sungkaew, S., Teerawatananon, A., Ohrnberger, D., Xia, N. H., Chan, K. S., et al. (2018). The hybrid origin of Phai Liang, a bamboo of recent introduction into horticulture in Southeast Asia, and a new nothogenus, xThyrsocalamus (Bambuseae: Bambusinae). Phytotaxa 362 (3), 271–281. doi: 10.11646/phytotaxa.362.3.3
Guo, C., Guo, Z. H., Li, D. Z. (2019). Phylogenomic analyses reveal intractable evolutionary history of a temperate bamboo genus (Poaceae: Bambusoideae). Plant Divers. 41 (4), 213–219. doi: 10.1016/j.pld.2019.05.003
Guo, Z. H., Ma, P. F., Yang, G. Q., Hu, J. Y., Liu, Y. L., Xia, E. H., et al. (2019). Genome sequences provide insights into the reticulate origin and unique traits of woody bamboos. Mol. Plant 12 (10), 1353–1365. doi: 10.1016/j.molp.2019.05.009
Guo, C., Ma, P. F., Yang, G. Q., Ye, X. Y., Guo, Y., Liu, J. X., et al. (2021). Parallel ddRAD and genome skimming analyses reveal a radiative and reticulate evolutionary history of the temperate bamboos. Syst. Biol. 70 (4), 756–773. doi: 10.1093/sysbio/syaa076
Haevermans, T., Mantuano, D., Zhou, M. Y., Lamxay, V., Haevermans, A., Blanc, P., et al. (2020). Discovery of the first succulent bamboo (Poaceae, Bambusoideae) in a new genus from Lao's karst areas, with a unique adaptation to seasonal drought. Phytokeys 156, 125–137. doi: 10.3897/phytokeys.156.51636
Huson, D. H., Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23 (2), 254–267. doi: 10.1093/molbev/msj030
Janzen, D. H. (1976). Why bamboos wait so long to flower. Annu. Rev. Ecol. Syst. 7, 347–391. doi: 10.1146/annurev.es.07.110176.002023
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Johnson, M. G., Gardner, E. M., Liu, Y., Medina, R., Goffinet, B., Shaw, A. J., et al. (2016). HybPiper: Extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl. Plant Sci. 4 (7), 1600016. doi: 10.3732/apps.1600016
Kambale, N., Hegbe, A. D. M. T., Minengu Mayulu, J. D., Muhindo Sahani, W., Biaou, S. S. H., Natta, A. K. (2022). Bamboos (Bambusiadeae): plant resources with ecological, socio-economic and cultural virtues: a review. J. Environ. Sci. 21 (4), 1–14.
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 (12), 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelchner, S. A., Bamboo Phylogeny Group (2013). Higher level phylogenetic relationships within the bamboos (Poaceae: Bambusoideae) based on five plastid markers. Mol. Phylogenet. Evol. 67 (2), 404–413. doi: 10.1016/j.ympev.2013.02.005
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Li, D. Z., Stapleton, C. M. A. (2006). Neomicrocalamus P.C. Keng, in Flora of China Vol. 22. Eds. Wu, Z. Y., Raven, P. H. (Beijing, China and Saint Louis, USA: Science Press and Missouri Botanical Garden Press), 57.
Linder, C. R., Rieseberg, L. H. (2004). Reconstructing patterns of reticulate evolution in plants. Am. J. Bot. 91 (10), 1700–1708. doi: 10.3732/ajb.91.10.1700
Liu, B. B., Ma, Z. Y., Ren, C., Hodel, R. G. J., Sun, M., Liu, X. Q., et al. (2021). Capturing single-copy nuclear genes, organellar genomes, and nuclear ribosomal DNA from deep genome skimming data for plant phylogenetics: a case study in Vitaceae. J. Syst. Evol. 59 (5), 1124–1138. doi: 10.1111/jse.12806
Liu, J. X., Xu, Z. C., Zhang, Y. X., Zhou, M. Y., Li, D. Z. (2022). The identity of Dinochloa species and enumeration of Melocalamus (Poaceae: Bambusoideae) in China. Plant Divers. 45 (2), 133–146. doi: 10.1016/j.pld.2022.07.001
Liu, J. X., Zhou, M. Y., Yang, G. Q., Zhang, Y. X., Ma, P. F., Guo, C., et al. (2020). ddRAD analyses reveal a credible phylogenetic relationship of the four main genera of Bambusa-Dendrocalamus-Gigantochloa complex (Poaceae: Bambusoideae). Mol. Phylogenet. Evol. 146, 106758. doi: 10.1016/j.ympev.2020.106758
Malinsky, M., Matschiner, M., Svardal, H. (2021). Dsuite-Fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 21 (2), 584–595. doi: 10.1111/1755-0998.13265
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A., Zdobnov, E. M. (2021). BUSCO Update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38 (10), 4647–4654. doi: 10.1093/molbev/msab199
Marçais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinform 27, 764–770. doi: 10.1093/bioinformatics/btr011
McCauley, D. E. (1994). Contrasting the distribution of chloroplast DNA and allozyme polymorphism among local populations of Silene alba: implications for studies of gene flow in plants. PNAS 91 (17), 8127–8131. doi: 10.1073/pnas.91.17.8127
McCormack, J. E., Huang, H., Knowles, L. L. (2009). Maximum likelihood estimates of species trees: how accuracy of phylogenetic inference depends upon the divergence history and sampling design. Syst. Biol. 58 (5), 501–508. doi: 10.1093/sysbio/syp045
Morales-Briones, D. F., Liston, A., Tank, D. C. (2018). Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the neotropical genus Lachemilla (Rosaceae). New Phytol. 218 (4), 1668–1684. doi: 10.1111/nph.15099
Mugrabi De Kuppler, A. L., Fagúndez, J., Bellstedt, D. U., Oliver, E. G. H., Léon, J., Pirie, M. D. (2015). Testing reticulate versus coalescent origins of Erica lusitanica using a species phylogeny of the northern heathers (Ericeae, Ericaceae). Mol. Phylogenet. Evol. 88, 121–131. doi: 10.1016/j.ympev.2015.04.005
Nguyen, T. Q. (1991). A new genus and the new speceis of bamboos (Poaceae, Bambusoideae) from Vietnam. Bot. Zhurn. (Moscow Leningrad) 76 (6), 874–880.
Nylander, J. A. A. (2004). MrModeltest v2. Program distributed by the author (Uppsala, Sweden: Evolutionary Biology Centre, Uppsala University).
Ortiz, E. M. (2019). vcf2phylip v2.0: convert a VCF matrix into several matrix formats for phylogenetic analysis. (v2.0). Zenodo. doi: 10.5281/zenodo.2540861
Pease, J. B., Hahn, M. W. (2015). Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64 (4), 651–662. doi: 10.1093/sysbio/syv023
Petit, R. J., Excoffier, L. (2009). Gene flow and species delimitation. Trends Ecol. Evol. 24 (7), 386–393. doi: 10.1016/j.tree.2009.02.011
Posada, D., Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53 (5), 793–808. doi: 10.1080/10635150490522304
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi: 10.1086/519795
Qu, X. J., Moore, M. J., Li, D. Z., Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 50. doi: 10.1186/s13007-019-0435-7
Ranallo-Benavidez, T. R., Jaron, K. S., Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432. doi: 10.1038/s41467-020-14998-3
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Ruiz-Sanchez, E., Sosa, V. (2015). Origin and evolution of fleshy fruit in woody bamboos. Mol. Phylogenet. Evol. 91, 123–134. doi: 10.1016/j.ympev.2015.05.020
Scheunert, A., Heubl, G. (2017). Against all odds: reconstructing the evolutionary history of Scrophularia (Scrophulariaceae) despite high levels of incongruence and reticulate evolution. Org. Divers. Evol. 17 (2), 323–349. doi: 10.1007/s13127-016-0316-0
Smith, S. A., Moore, M. J., Brown, J. W., Yang, Y. (2015). Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15, 150. doi: 10.1186/s12862-015-0423-0
Solís-Lemus, C., Ané, C. (2016). Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PloS Genet. 12 (3), e1005896. doi: 10.1371/journal.pgen.1005896
Solís-Lemus, C., Bastide, P., Ané, C. (2017). PhyloNetworks: a package for phylogenetic networks. Mol. Biol. Evol. 34 (12), 3292–3298. doi: 10.1093/molbev/msx235
Soltis, P. S., Folk, R. A., Soltis, D. E. (2019). Darwin review: angiosperm phylogeny and evolutionary radiations. Proc. R. Soc B: Biol. Sci. 286, 20190099. doi: 10.1098/rspb.2019.0099
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stapleton, C. M. A. (1994). The bamboos of Nepal and Bhutan. part III: Drepanostachyum, Himalayacalamus, Ampelocalamus, Neomicrocalamus and Chimonobambusa (Gramineae: Poaceae, Bambusoideae). Edinb. J. Bot. 51 (3), 301–330. doi: 10.1017/S0960428600001815
Stapleton, C. M. A., Li, D. Z., Xue, J. R. (1997). A new combination in Cephalostachyum with notes on names in Neomicrocalamus (Gramineae: Bambusoideae). Kew Bull. 52 (3), 699–702. doi: 10.2307/4110299
Stull, G. W., Soltis, P. S., Soltis, D. E., Gitzendanner, M. A., Smith, S. A. (2020). Nuclear phylogenomic analyses of asterids conflict with plastome trees and support novel relationships among major lineages. Am. J. Bot. 107 (5), 790–805. doi: 10.1002/ajb2.1468
Sukumaran, J., Holder, M. T. (2010). DendroPy: a Python library for phylogenetic computing. Bioinformatics 26 (12), 1569–1571. doi: 10.1093/bioinformatics/btq228
Sungkaew, S., Stapleton, C. M. A., Salamin, N., Hodkinson, T. R. (2009). Non-monophyly of the woody bamboos (Bambuseae; Poaceae): a multi-gene region phylogenetic analysis of Bambusoideae s.s. J. Plant Res. 122 (1), 95–108. doi: 10.1007/s10265-008-0192-6
Swofford, D. L. (2003). PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4 (Sunderland, UK: Sinauer Associates).
van der Auwera, G., O'Connor, B. D. (2020). Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (California, USA: O'Reilly Media, Incorporated).
Vorontsova, M. S., Clark, L. G., Dransfield, J., Govaerts, R., Baker, W. J. (2016). World Checklist of Bamboos and Rattans (Beijing, China: International Network for Bamboo and Rattan and Kew, UK: Royal Botanic Gardens, Kew).
Wang, X. Q., Ye, X. Y., Zhao, L., Li, D. Z., Guo, Z. H., Zhuang, H. F. (2017). Genome-wide RAD sequencing data provide unprecedented resolution of the phylogeny of temperate bamboos (Poaceae: Bambusoideae). Sci. Rep. 7, 11546. doi: 10.1038/s41598-017-11367-x
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31 (20), 3350–3352. doi: 10.1093/bioinformatics/btv383
Wong, K. M., Goh, W. L., Chokthaweepanich, H., Clark, L. G., Sungkaew, S., Widjaja, E. A., et al. (2016). A subtribal classifcation of Malesian and Southwest Pacifc woody bamboos (Poaceae: Bambusoideae: Bambuseae) informed by morphological and molecular studies. Sandakania 22, 11–36.
Xu, H. B., Luo, X., Qian, J., Pang, X. H., Song, J. Y., Qian, G. R., et al. (2012). FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7 (12), e52249. doi: 10.1371/journal.pone.0052249
Ye, X. Y., Ma, P. F., Guo, C., Li, D. Z. (2021). Phylogenomics of Fargesia and Yushania reveals a history of reticulate evolution. J. Syst. Evol. 59 (6), 1183–1197. doi: 10.1111/jse.12719
Zhang, C., Rabiee, M., Sayyari, E., Mirarab, S. (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19 (S6), 153. doi: 10.1186/s12859-018-2129-y
Zhang, Y. X., Zeng, C. X., Li, D. Z. (2012). Complex evolution in Arundinarieae (Poaceae: Bambusoideae): Incongruence between plastid and nuclear GBSSI gene phylogenies. Mol. Phylogenet. Evol. 63 (3), 777–797. doi: 10.1016/j.ympev.2012.02.023
Zhao, H. S., Gao, Z. M., Wang, L., Wang, J. L., Wang, S. B., Fei, B. H., et al. (2018). Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). GigaScience 7, 1–12. doi: 10.1093/gigascience/giy115
Zheng, Y. S., Yang, D. M., Rong, J. D., Chen, L. G., Zhu, Q., He, T. Y., et al. (2022). Allele-aware chromosome-scale assembly of the allopolyploid genome of hexaploid Ma bamboo (Dendrocalamus latiflorus Munro). J. Integr. Plant Biol. 64 (3), 649–670. doi: 10.1111/jipb.13217
Zhou, Y., Duvaux, L., Ren, G., Zhang, L., Savolainen, O., Liu, J. (2017). Importance of incomplete lineage sorting and introgression in the origin of shared genetic variation between two closely related pines with overlapping distributions. Hered 118 (3), 211–220. doi: 10.1038/hdy.2016.72
Zhou, B. F., Yuan, S., Crowl, A. A., Liang, Y. Y., Shi, Y., Chen, X. Y., et al. (2022). Phylogenomic analyses highlight innovation and introgression in the continental radiations of Fagaceae across the Northern Hemisphere. Nat. Commun. 13, 1320. doi: 10.1038/s41467-022-28917-1
Keywords: single-nucleotide polymorphism, single-copy nuclear gene, phylogenetic incongruence, introgression, incomplete lineage sorting
Citation: Cai Z-Y, Niu Z-Y, Zhang Y-Y, Tong Y-H, Vu TC, Goh WL, Sungkaew S, Teerawatananon A and Xia N-H (2023) Phylogenomic analyses reveal reticulate evolution between Neomicrocalamus and Temochloa (Poaceae: Bambusoideae). Front. Plant Sci. 14:1274337. doi: 10.3389/fpls.2023.1274337
Received: 08 August 2023; Accepted: 14 November 2023;
Published: 04 December 2023.
Edited by:
Xiaohua Jin, Institute of Botany, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Loreta Brandão de Freitas, Federal University of Rio Grande do Sul, BrazilChristopher Tyrrell, Milwaukee Public Museum, United States
Eduardo Ruiz-Sanchez, University of Guadalajara, Mexico
Copyright © 2023 Cai, Niu, Zhang, Tong, Vu, Goh, Sungkaew, Teerawatananon and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nian-He Xia, nhxia@scbg.ac.cn