 Xue-Min Xu
Xue-Min Xu Zhen Wei
Zhen Wei Jun-Zhe Sun
Jun-Zhe Sun Yang Lu
Yang Lu Zhen-Long Wang
Zhen-Long Wang Shi-Xin Zhu
Shi-Xin Zhu- School of Life Sciences, Zhengzhou University, Zhengzhou, China
The infrageneric taxonomy system, species delimitation, and interspecies systematic relationships of Leontopodium remain controversial and complex. However, only a few studies have focused on the molecular phylogeny of this genus. In this study, the characteristics of 43 chloroplast genomes of Leontopodium and its closely related genera were analyzed. Phylogenetic relationships were inferred based on chloroplast genomes and nuclear ribosomal DNA (nrDNA). Finally, together with the morphological characteristics, the relationships within Leontopodium were identified and discussed. The results showed that the chloroplast genomes of Filago, Gamochaeta, and Leontopodium were well-conserved in terms of gene number, gene order, and GC content. The most remarkable differences among the three genera were the length of the complete chloroplast genome, large single-copy region, small single-copy region, and inverted repeat region. In addition, the chloroplast genome structure of Leontopodium exhibited high consistency and was obviously different from that of Filago and Gamochaeta in some regions, such as matk, trnK (UUU)-rps16, petN-psbM, and trnE (UUC)-rpoB. All the phylogenetic trees indicated that Leontopodium was monophyletic. Except for the subgeneric level, our molecular phylogenetic results were inconsistent with the previous taxonomic system, which was based on morphological characteristics. Nevertheless, we found that the characteristics of the leaf base, stem types, and carpopodium base were phylogenetically correlated and may have potential value in the taxonomic study of Leontopodium. In the phylogenetic trees inferred using complete chloroplast genomes, the subgen. Leontopodium was divided into two clades (Clades 1 and 2), with most species in Clade 1 having herbaceous stems, amplexicaul, or sheathed leaves, and constricted carpopodium; most species in Clade 2 had woody stems, not amplexicaul and sheathed leaves, and not constricted carpopodium.
1 Introduction
Leontopodium R.Br. ex Cass. (Asteraceae) comprises 58 species that are distributed across Asia and Europe (Bayer et al., 2007; Chen et al., 2011). The main distribution of the genus is in central and eastern Asia, including Russia, Japan, South Korea, Mongolia, China and along the Himalaya to the borders of Afghanistan and Pakistan; two species occur in Europe, L. alpinum Cass. and L. nivale (Ten.) Huet ex Hand.-Mazz. (Safer et al., 2011). China has the largest number of Leontopodium species in the world, with a total of 37 species, of which 17 are endemic, and the West and Southwest regions of China are the diversity centers of this genus (ca. 20 species) (Lin, 1979; Chen et al., 2011). The genus is mainly characterized by capitula surrounded by bracteal leaves, involucral bracts with membranous margins, central florets that are functionally male, and pappi of bisexual flowers that are usually slightly thicker (Lin, 1979; Chen et al., 2011). Some taxa of this genus, such as L. artemisiifolium (Levl.) Beauv., L. calocephalum var. uliginosum Beauv., and L. leontopodioides (Willd.) Beauv. are commonly used as herbal remedies in China because of their antioxidant, anti-inflammatory, anti-rheumatic, and anti-diabetic properties (Lin, 1979; Jiao et al., 1997; Huang and Wu, 2006; Wu et al., 2013).
The phylogenetic position and closely related genera of Leontopodium are well defined. Traditionally, Leontopodium has been classified as belonging to the tribe Inuleae Cass. (Bentham, 1873; Cassini, 1822; Lin, 1965; Merxmüller et al., 1977; Lin, 1979). Subsequently, tribe Gnaphalieae Cass. ex Lecoq & Juill. was widely accepted based on morphological characteristics and molecular phylogenetic results, with Leontopodium belonging to it (Anderberg, 1989; Jansen et al., 1991; Anderberg, 1991a; Anderberg, 1991b; Anderberg, 1991c; Karis, 1993; Bayer and Starr, 1998; Eldenäs et al., 1999; Bayer et al., 2007; Galbany-Casals et al., 2010; Fu et al., 2016; Nie et al., 2016; Smissen et al., 2020). The latest research indicated that Gnaphalieae were one of the larger tribes of Asteraceae with c. 2,100 species in 178 genera, occurring globally across a wide range of temperate habitats (Smissen et al., 2020). Based on rpl32-trnL, trnL intron, trnL-trnF, and the nuclear ribosomal DNA internal transcribed spacer (ITS) and external transcribed spacer (ETS), Galbany-Casals et al. (2010) reported that Leontopodium had a close relationship with Antennaria Gaertn., Bombycilaena (DC.) Smoljan., Gamochaeta Wedd., Evax Gaertn., Filago Loefl., etc.; thus, the “FLAG clade” was proposed (Filago, Leontopodium, Antennaria, and Gamochaeta are the largest genera in this clade). In addition, the results of previous molecular phylogenetic studies support the monophyly of Leontopodium. Nie et al. (2016) explored the phylogenetic relationships within Gnaphalieae based on abundant samples (a total of 835 terminal accessions representing 80% of the genera, including 27 Leontopodium species) and using ITS and ETS sequences; their results supported the “FLAG clade” and indicated that Leontopodium was monophyletic. Smissen et al. (2020) suggested a subdivision of Gnaphalieae into two subtribes based on published studies and their new phylogenetic analyses; these were a largely African-endemic Relhaniinae (124 species in 11 genera) and a much enlarged Gnaphaliinae, the latter accounting for more than 90% of the species diversity (c. 2,000 species in 167 genera) and including six clades, Ifloga, Metalasia, Stoebe, HAP, FLAG (Leontopodium was in this clade and monophyletic), and Australasian clades.
However, the infrageneric taxonomy system, species delimitation, and interspecies systematic relationships of Leontopodium remain controversial and complex. Handel-Mazzetti (1928) accepted 41 Leontopodium species and divided the genus into two subgenera (subgen. Paragnaphalium Hand.-Mazz. and subgen. Euleontopodium Beauv.), and two sections (sect. Nobilia (Beauv.) Hand.-Mazz. and sect. Alpina Hand.-Mazz.); only one species was included in subgen. Paragnaphalium (L. forrestianum Hand.-Mazz.). Blöch et al. (2010) treated several of the taxa proposed by Handel-Mazzetti (1928) as synonyms and pointed out that Leontopodium may comprise only 30 species. Lin (1965) indicated the existence of about 40 species and 12 natural hybrids of Leontopodium in China and divided this genus into two subgenera (subgen. Paragnaphalium and subgen. Leontopodium), two sections (sect. Nobilia and sect. Leontopodium), eight subsections, and 12 series. Whereas Chen et al. (2011) revised Leontopodium in China and indicated that there were only 37 species in this genus; moreover, they did not set up an infrageneric taxonomy system and did not accept hybrids, because it was somewhat difficult to distinguish them according to the knowledge available at that time. To date, only two studies have focused on the molecular phylogeny of some Leontopodium species. Blöch et al. (2010) explored the relationships of 22 Leontopodium species using three chloroplast markers (matK, trnL intron, and trnL-trnF) and two nuclear genes (ITS and ETS); their results showed that the Southeast Tibetan monotypic Sinoleontopodium (S. lingianum Y.L. Chen) fell into Leontopodium, and L. lingianum (Y.L. Chen) Dickoré, comb. nov., was proposed, to ensure the monophyly of Leontopodium. Safer et al. (2011) divided 16 Leontopodium species into 10 groups according to the results of Amplified Fragment Length Polymorphism, to discuss the relationships within the genus and to reveal information about its biogeography. Thus, few taxa were included, and few molecular markers have been used in previous phylogenetic studies of Leontopodium, which has led to incomplete and indistinct interspecies phylogenetic relationships. Accordingly, additional molecular data and taxa should be used to investigate the interspecies relationships of Leontopodium.
Phylogenetic analyses using chloroplast (cp) and nuclear genes are more comprehensive and can be used to explore complex genetic relationships. The cp, which has independent genetic material (mainly maternally inherited), is responsible for photosynthesis and plays important roles in other aspects of plant physiology and development, including the synthesis of various proteins, nucleotides, carbohydrates, and metabolites (Leister, 2003; Xiong et al., 2009; Wicke et al., 2011; Daniell et al., 2016; Tian et al., 2021). Generally, the cp genome has a typical quadripartite circular structure comprising two copies of inverted repeat (IR) regions, a large single-copy (LSC) region, and a small single-copy (SSC) region (Palmer, 1985; Qian et al., 2021). In addition, cp genomes are highly conserved, not only in structure, but also in gene number and composition, usually ranging from 120 to 220 kb and including 120–130 genes (Jansen et al., 2008; Rogalski et al., 2015). Moreover, the evolutionary rate of cp genomes is relatively moderate and lies between those of the nuclear and mitochondrial genomes (Dong et al., 2013). Owing to the lack of recombination, small genome size, and high copy number per cell, complete cp genome sequences have significantly contributed to phylogenetic studies and plant classification (Dong et al., 2012; Twyford and Ness, 2017; Jiang et al., 2020; Pascual-Díaz et al., 2021; Sun et al., 2021; Wang et al., 2021; Zhang X. F. et al., 2021). Mutation hotspot regions and single-sequence repeats can be identified by comparing cp genome sequences and are commonly used as effective molecular markers for species identification and classification, population genetics, and evolutionary studies (Dong et al., 2012). Furthermore, because the variation in genome structure is often considered a type of evolutionary event in general (Jansen and Ruhlman, 2012), the differences in the cp genome structure are often analyzed in great detail among species (Jansen et al., 2006; Liu et al., 2013; Barkalov and Kozyrenko, 2014; Lee et al., 2016; Ross et al., 2016; Ng et al., 2017; Sam et al., 2017; Wei et al., 2017; Wu et al., 2017; Zhang et al., 2017; Yang et al., 2018; Jiang et al., 2020; Sun et al., 2021). Currently, the National Center for Biotechnology Information (NCBI) database includes many complete cp genomes of Asteraceae (approximately 1760) (https://www.ncbi.nlm.nih.gov/). However, to date, only one complete cp genome of Leontopodium (L. leiolepis Nakai, KM267636) has been published in the NCBI database. Therefore, additional cp genome data must be analyzed to unveil the infrageneric taxonomic system, species delimitation, and interspecies systematic relationships of Leontopodium. Compared with genomic data, phylogenetic analysis based on DNA barcodes, such as trnL-F, rbcL, matK, trnK-matK, psbA-trnH, ITS, and ETS, can solve many taxonomic problems, among which ITS and ETS are important nuclear DNA fragments that are widely used in phylogenetic studies (Kuzmanović et al., 2017; Malekmohammadi et al., 2017; Vicent et al., 2017; Zhou and Zhang, 2017; García et al., 2018; Asanuma et al., 2019; Hussain et al., 2019; Pirani et al., 2020; Tan et al., 2020; Zhou et al., 2020; Hashim et al., 2021). ITS and ETS have fast evolutionary rates and high interspecies variation, which are reportedly effective in discriminating closely related species with relatively recent divergences (Li et al., 2011). In addition, incongruent genetic relationships among the different topological trees constructed using cp and nuclear genes suggest the existence of hybridization, incomplete lineage sorting, and/or interspecific introgression (Yang et al., 2013; Gatesy et al., 2019), and complex genetic relationships can be revealed by phylogenetic analyses based on cp and nuclear genes. However, few studies have focused on the infrageneric taxonomic system, species delimitation, and interspecies systematic relationships of Leontopodium based on cp and nuclear genes.
Therefore, in this study, the structure, gene content, and general characteristics of 43 cp genomes were compared and analyzed in detail to explore the evolution of the cp genome. Subsequently, phylogenetic trees were constructed using cp genomes and nrDNA to identify markers that are more effective for phylogenetic resolution, examine the phylogenetic relationships within Leontopodium from China, and further determine the phylogenetic position of Leontopodium. Finally, together with morphological characteristics, the infrageneric taxonomy system, species delimitation, and interspecies systematic relationships of Leontopodium were identified and discussed.
2 Materials and methods
2.1 Taxon sampling
The sequences used in this study included both new sequences and previously published sequences. Leaf materials of 43 individuals representing 30 taxa (one Filago, three Gamochaeta, and 26 Leontopodium) were obtained. Most materials used in this study were collected from natural populations in China, and voucher specimens were deposited in the herbarium of Zhengzhou University (ZZU; Zhengzhou, China). Leaf materials of a few taxa were obtained from herbarium specimens of PE (Institute of Botany, Chinese Academy of Sciences, Beijing, China). Detailed information on the samples is provided in Table 1. The NCBI database accession numbers of the new sequences (cp genomes, ITS, and ETS) in this study are shown in Table 2. Furthermore, the cp genome of Gamochaeta coarctata Kerguélen (MK570596) was acquired for phylogenetic reconstruction. Previously published nrDNA sequences (205 ITS and 205 ETS sequences of 101 taxa, including four Antennaria, 33 Filago, 30 Gamochaeta, and 34 Leontopodium) of the same individuals were obtained from the NCBI database and are listed in Supplementary Table 1. Based on phylogenetic results reported by Fu et al. (2016); Huang et al. (2016); Panero and Crozier (2016); Mandel et al. (2019) and Zhang C. et al. (2021), Calendula arvensis M.Bieb. was used as an outgroup (NCBI accession numbers: cp genome, ON641308; ITS, GU818507; and ETS, GU818129).
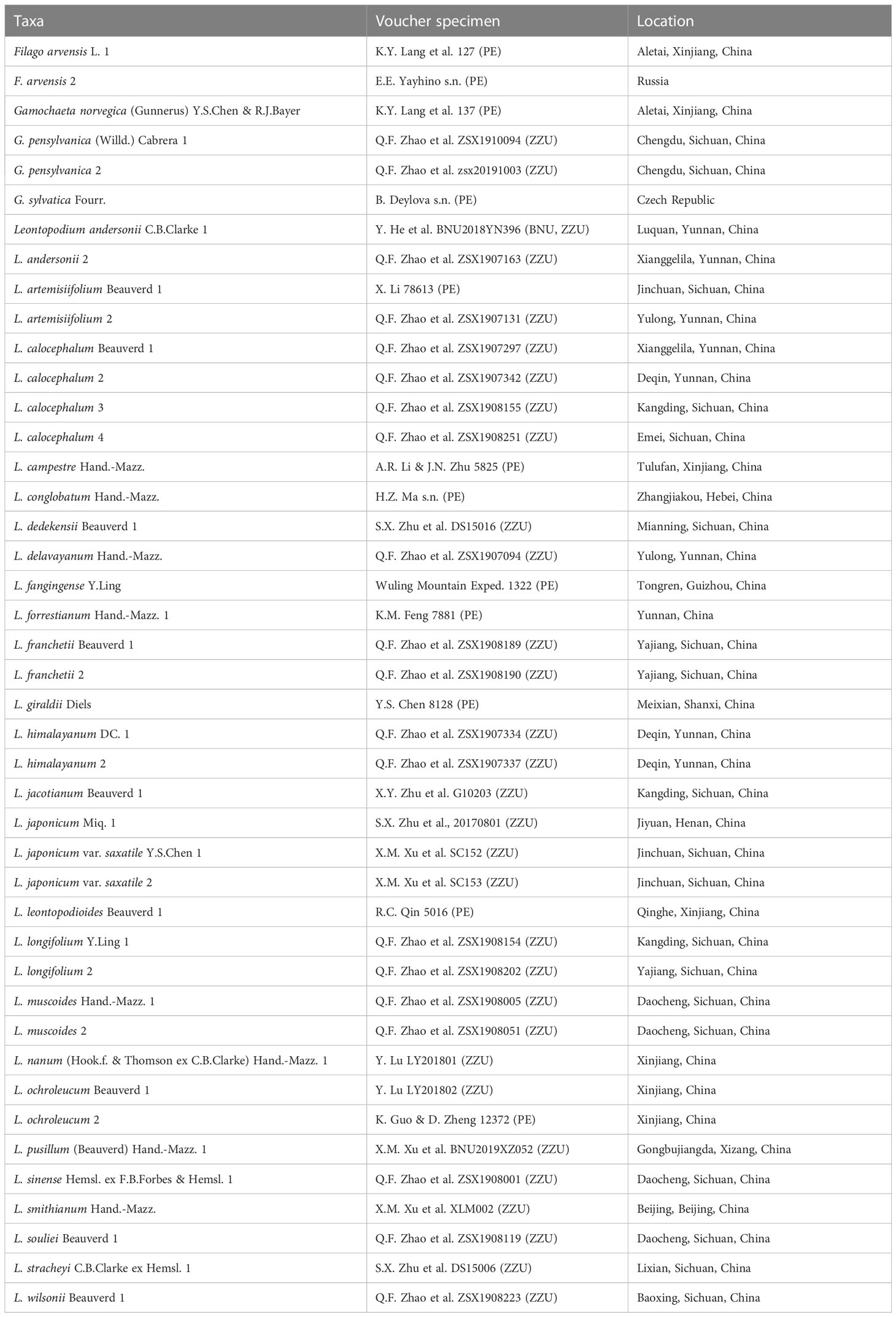
Table 1 Voucher specimens and location information.
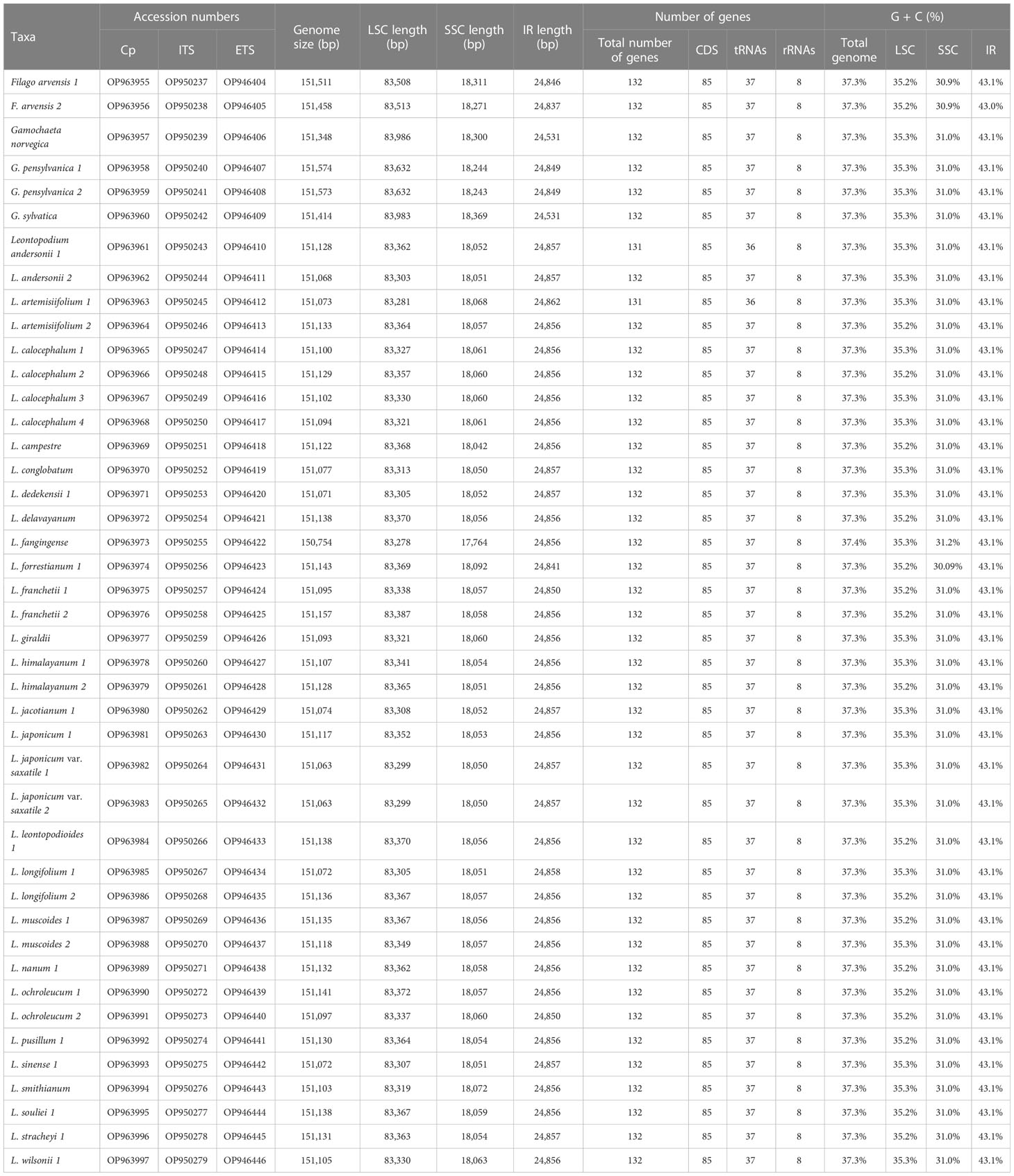
Table 2 Accession numbers of new sequences (chloroplast genomes, ITS, and ETS) and the features of chloroplast genomes.
2.2 DNA extraction, genome sequencing, and assembly
We used a modified cetyltrimethylammonium bromide (CTAB) method to extract high-quality DNA (Doyle and Doyle, 1987), which was then purified using the Wizard® DNA cleanup system (Promega, Madison, WI, USA). DNA quality was assessed using a NanoDrop spectrophotometer (Thermo Scientific, Carlsbad, CA, USA), and DNA integrity was evaluated by electrophoresis on a 1% (w/v) agarose gel. A DNA library was prepared using the NEB Next Ultra DNA Library Prep Kit for Illumina (San Diego, CA, USA). Libraries for paired-end 150-bp sequencing were analyzed on an Illumina NovaSeq 6000 platform (Novogene Co., Ltd., Tianjin, China) to generate approximately 10 GB of data for each sample. Raw reads were filtered using SOAPnuke to remove sequencing adaptors and low-quality bases (Chen et al., 2018). The filtered reads were assembled using GetOrganelle (Jin et al., 2020) with a range of 21, 45, 65, 85, and 105 k-mers for plastomes and 35, 85, and 115 k-mers for nrDNA. Subsequently, ITS and ETS sequences were uploaded to the NCBI GenBank database (accession numbers are listed in Table 2).
2.3 Cp genome annotation and comparative analysis
The plastome sequences were initially annotated using Geneious Prime 2020.1.2 (https://www.geneious.com) by referring to the cp genome sequence of Anaphalis sinica Hance (KX148081), Anaphalis margaritacea var. yedoensis Ohwi (LC656264), and G. coarctata (MK570596). Annotations of protein-coding sequences were manually checked based on the open reading frame. Transfer RNA (tRNA) genes were verified using the online tRNAscan-SE tool with default settings (Lowe and Chan, 2016). All cp genome sequences have been deposited in the NCBI GenBank database (accession numbers are listed in Table 2). The complete cp genome was visualized using OGDRAW (Greiner et al., 2019). The mVISTA program in Shuffle-LAGAN mode was used to compare the cp genomes, using Filago arvensis L. (OP963955) as the reference (Frazer et al., 2004). The junctions and borders of the IR regions were visualized using IRScope (Amiryousefi et al., 2018). DnaSP version 6 was used to calculate the nucleotide variability (Pi) among the cp genomes (Rozas et al., 2017).
2.4 Phylogenetic analysis
Phylogenetic topology was constructed based on five matrices: complete cp sequences, coding genes of chloroplast genomes, ITS, ETS, and concatenated sequences of ITS and ETS. An online version of MAFFT (Katoh et al., 2019) was used to align the datasets. Phylogenetic analyses were performed using maximum likelihood (ML) and Bayesian inference (BI) methods using IQ-TREE v1.6.12 and MrBayes 3.2.2, respectively (Ronquist et al., 2012; Nguyen et al., 2015). The best-fitting model of nucleotide substitutions was determined using ModelFinder in PhyloSuite v1.2.2 (Zhang D. et al., 2020). ML analyses were performed using IQ-TREE with 1,000 bootstrap (BS) replicates. BI analysis was run for 5,000,000 generations and sampled every 5,000 generations; the first 25% of the trees were discarded as burn-in. Trees were selected based on a 50% majority-rule consensus to estimate posterior probabilities (PP). The effective sample size (>200) was determined using Tracer v1.7 (Rambaut et al., 2018). The reconstructed trees were visualized using Figtree V.1.4.2 (Rambaut, 2014) and TreeGraph 2 (Stöver and Müller, 2010).
3 Results
3.1 Characteristics of cp genomes
A total of 43 cp genomes (30 taxa, including one Filago, three Gamochaeta, and 26 Leontopodium) were compared. All cp genomes had a typical quadripartite structure: an LSC, SSC, and two IRs (Figure 1). Among the 43 samples, the total length of cp genomes ranged from 150,754 bp (L. fangingense, OP963973) to 151,574 bp (G. pensylvanica, OP963958) (Table 2). The lengths of LSC, SSC, and IRs ranged from 83,278 bp (L. fangingense) to 83,986 bp (G. norvegica, OP963957); 17,764 bp (L. fangingense) to 18,369 bp (G. sylvatica, OP963960); and 24,531 bp (G. norvegica) to 24,862 bp (L. artemisiifolium, OP963963), respectively (Table 2). The lengths of the complete cp genomes, LSC, SSC, and IRs of Leontopodium ranged from 150,754 bp (L. fangingense) to 151,157 bp (L. franchetii, OP963976), 83,278 bp (L. fangingense) to 83,387 bp (L. franchetii), 17,764 bp (L. fangingense) to 18,092 bp (L. forrestianum, OP963974), and 24,850 bp (L. franchetii) to 24,862 bp (L. artemisiifolium), respectively (Table 2). Compared with Filago and Gamochaeta, Leontopodium had longer whole cp genomes and LSC and SSC regions; however, the length of the IRs was slightly shorter. In Leontopodium, the length of the SSC region varied more significantly (328 bp) than that of the LSC (109 bp) and IR (21 bp) regions. The cp genomes comprised 131–132 genes, including 85 protein-coding, eight rRNA, and 36–37 tRNA genes (Table 2), two of which, L. artemisiifolium 1 and L. andersonii 1, lacked the trnT-GGU gene. The total GC content of the cp genomes was highly similar (37.3%–37.4%). The GC content of IRs (43.0%–43.1%) was higher than that of the LSC (35.2%–35.3%) and SSC (30.09%–31.2%) regions (Table 2). Compared with other species of Leontopodium, the GC content of the total cp genome and SSC region in L. fangingense was higher.
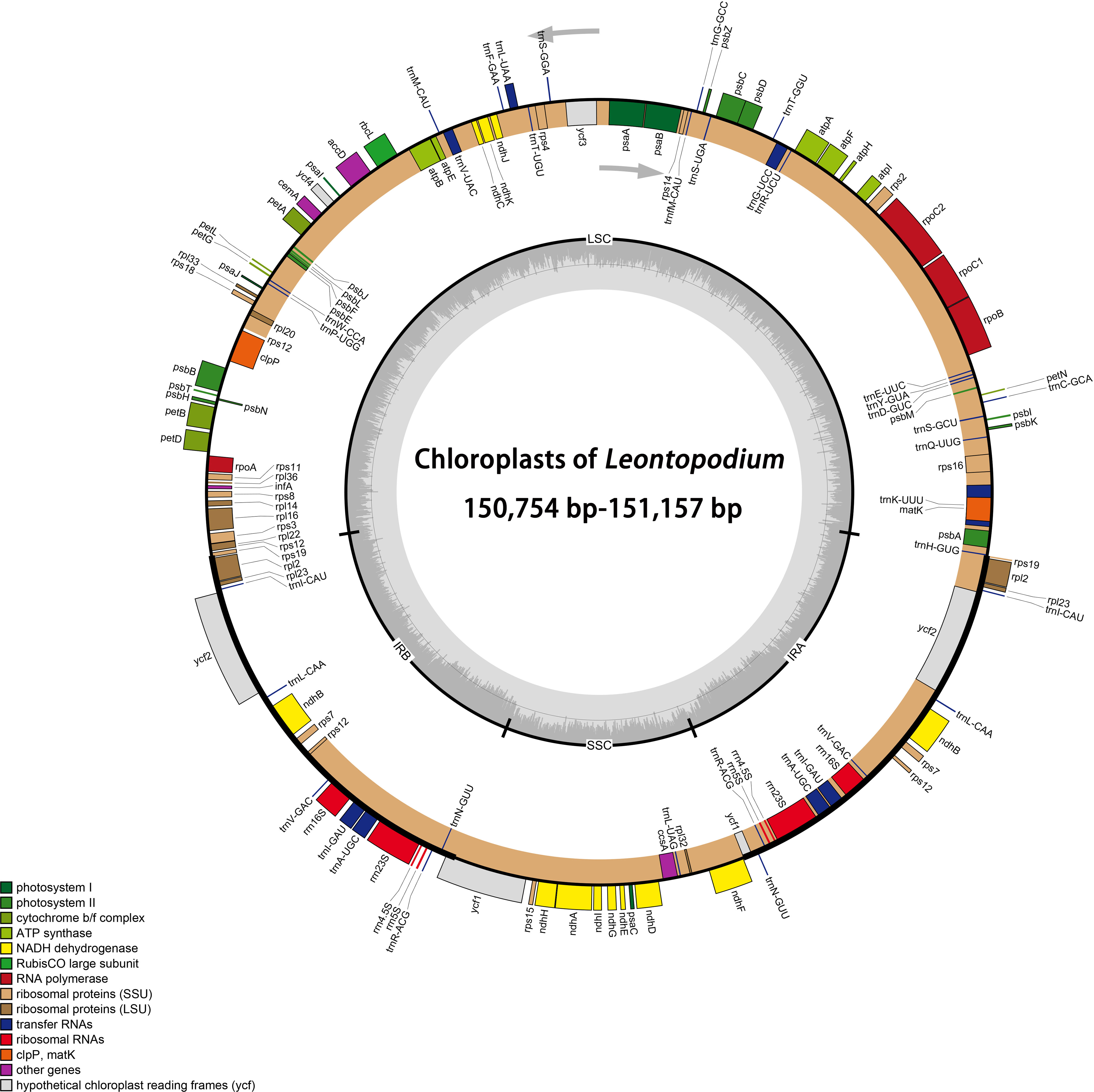
Figure 1 Chloroplast genome gene map of Leontopodium. Gray arrows indicate the direction of gene transcription. Genes belonging to different functional groups are marked with different colors. The dashed area in the inner circle indicates the GC content of the chloroplast genome; small single copy (SSC), large single copy (LSC), and inverted repeats (IRA and IRB) are indicated.
3.2 Boundaries between IR and SC regions
All 43 cp genomes were analyzed, and the differences among the junctions of the LSC/IRb (JLB), IRb/SSC (JSB), SSC/IRa (JSA), and IRa/LSC (JLA) regions were compared (Figure 2). Most cp genomes have similar characteristics. The junctions of the LSC/IRb regions in all the 43 cp genomes were located at rps19. Other than L. stracheyi, all taxa had 190 bp of rps19 in the LSC region and 89 bp in the IRb region. ycf1 was located at the IRb/SSC junction in all samples. F. arvensis had 564 bp ycf1 in the IRb region and 4,565–4,568 bp in the LSC region. The taxa of Gamochaeta had 460–564 bp ycf1 in the IRb region and 4,562–4,672 bp in the LSC region. In Leontopodium, except for L. forrestianum and L. himalayanum 2, all samples had 580 bp of ycf1 in the IRb regions and 4537 bp in the LSC regions. L. forrestianum had 564 bp of ycf1 in the IRb region and 4,553 bp in the LSC region. L. himalayanum 2 had 580 bp of ycf1 in the IRb region and 4,531 bp in the LSC region. The ndhF and ψycf1 genes were detected at the SSC/IRa boundary. Except for L. andersonii 1, the ndhF of all samples was located entirely in the SSC region. L. andersonii 1 had 29 bp of ndhF in the IRa region. The ψycf1 gene of some samples, such as G. norvegica, G. sylvatica, L. artemisiifolium 2, L. conglobatum, L. delavayanum, and L. japonicum, crossed the boundary of the SSC and IRa regions, with 4, 5, or 8 bp extending into the SSC region. The ψrps19 and trnH genes were detected in the IRa/LSC junctions of all 43 cp genomes.
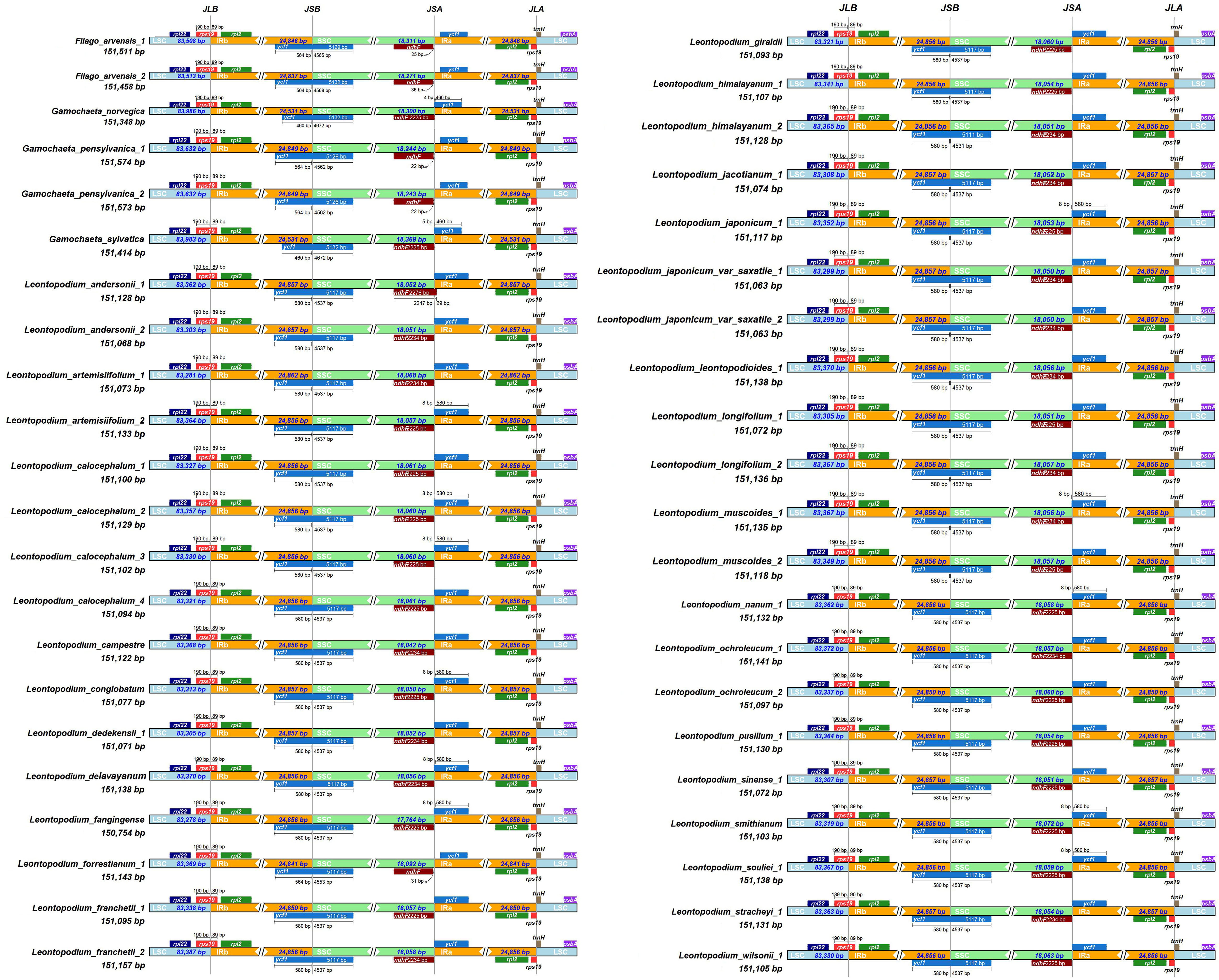
Figure 2 Comparison of the LSC, IR, and SSC border regions among the chloroplast genomes of Leontopodium, Filago, and Gamochaeta.
3.3 Comparative genomic analysis and divergence hotspot regions
The sequence divergence of the 43 cp genomes was comprehensively analyzed using the mVISTA program, with F. arvensis as a reference. Overall, the 43 cp genomes exhibited relatively high diversity (Figure 3), with genic regions being more conserved than intergenic spacer (IGS) regions. Most genic regions were highly convergent; the only divergent regions were detected in matK, atpA, rbcL, accD, rpoA, ycf1, ndhH, ndhG, and ndhF. The highest divergence was observed in IGS regions such as trnH (GUG)-psbA, trnK (UUU)-rps16, rps16-trnQ (UUG), petN-psbM, trnE (UUC)-rpoB, and atpI-atpH. The LSC and SSC regions showed a higher level of sequence divergence than the IR region. Moreover, the cp genome structure of Leontopodium exhibited higher consistency and was obviously different from that of Filago and Gamochaeta in some regions, such as matk, trnK (UUU)-rps16, rps16-trnQ (UUG), trnC (GCA)-petN, petN-psbM, and trnE (UUC)-rpoB. In Leontopodium, divergent regions were detected in the rps16-trnQ (UUG), trnE (UUC)-rpoB, ycf1-rps15, and clpP genes. Sliding window analyses of 43 cp genomes indicated that most of the variation occurred in the LSC and SSC regions, which exhibited high nucleotide variability (Figure 4A). In addition, 65 coding regions (aligned length >200 bp) and 88 non-coding regions (aligned length >200 bp) were extracted, and nucleotide variability was calculated (Supplementary Tables 2, 3). Moreover, the nucleotide diversity (Pi) values of the 65 coding regions and 88 non-coding regions were compared across the 43 cp genomes (Figures 4B, C). In the coding regions, the loci with the largest variation were ndhH, cemA, rps19, infA, psbH, atpB, rps15, ycf3, ndhJ, ndhF, ycf1, matK, and rbcL (Pi >0.002; Figure 4B); whereas in non-coding regions, the loci with the largest variation were trnK (UUU)-rps16, trnK (UUU)-matK, ndhD-ccsA, trnS (UGA)-psbZ, ndhI-ndhG, trnR (UCU)-trnG (UCC), rpl32-ndhF, trnM (CAU)-atpE, trnT (UGU)-trnL (UAA), petA-psbJ, trnL (UAG)-rpl32, ycf1-rps15, petN-psbM, and trnH (GUG)-psbA (Pi >0.006; Figure 4C).
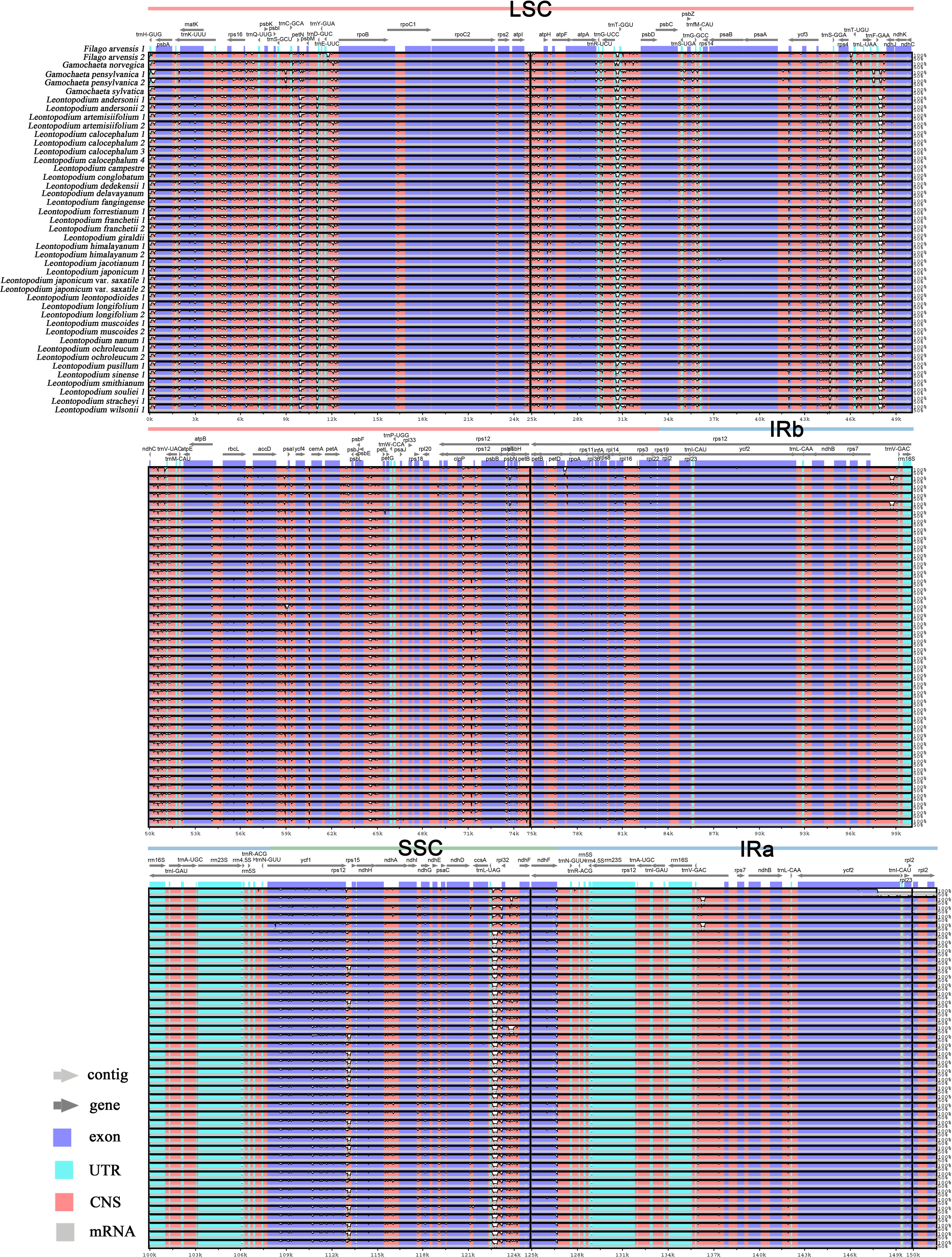
Figure 3 Sequence alignment of 43 chloroplast genomes using the mVISTA program, with F. arvensis as a reference. Genome regions are color-coded as protein-coding, rRNA-coding, tRNA-coding, or conserved noncoding sequences. The vertical scale indicates percentage identity, ranging from 50% to 100%. Regions with sequence variations among the chloroplast genomes are denoted in white.
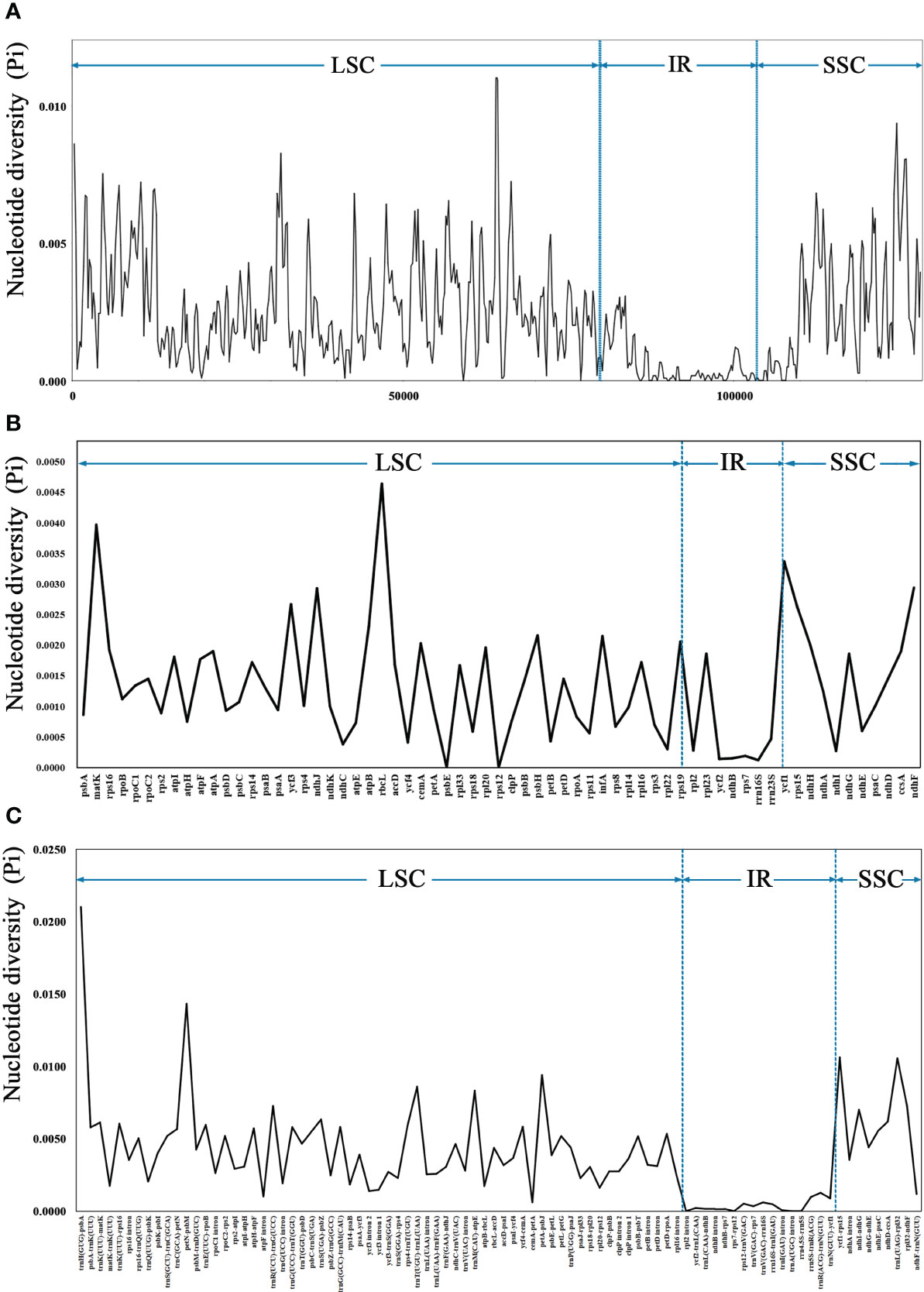
Figure 4 Nucleotide diversity (Pi) of 43 complete chloroplast genomes. (A) Sliding window analysis using a window length of 500 bp and step size of 200 bp. (B) Coding regions (aligned length >200 bp). (C) Noncoding regions (aligned length of >200 bp). The vertical dotted lines divide the approximate boundaries of LSC, IR, and SSC.
3.4 Phylogenetic results of cp genomes
The sequence characteristics and nucleotide substitution models for ML and BI phylogenetic analyses of different datasets (complete cp genomes, coding genes of chloroplast genomes, ITS, ETS, and concatenated sequences of ITS and ETS) are presented in Supplementary Table 4. In the phylogenetic trees that were inferred using complete cp genomes (Figure 5 and Supplementary Figure 1), Filago and Gamochaeta were sister groups and formed one clade, respectively, with strong support [Filago, ML bootstrap value (BS) = 100, Bayesian posterior probabilities (PP) = 1; Gamochaeta, BS = 72, PP = 0.98]. Leontopodium species were clustered into one clade (BS = 100, PP = 1) as a sister clade to the species to Filago and Gamochaeta species. In addition, three main clades were formed by the Leontopodium species (Figure 5 and Supplementary Figure 1). Clade 3 was a sister clade to Clade 1 and Clade 2, and included only one species, L. forrestianum. Clade 1 and Clade 2 included 17 and 10 taxa, respectively. However, in the phylogenetic trees constructed using complete cp genomes (Figure 5 and Supplementary Figure 1), the individuals of two species, L. artemisiifolium and L. longifolium, were in different main clades (Clade 1 and Clade 2). In Clade 1, the individuals of L. franchetii, L. himalayanum, L. muscoides, and L. ochroleucum, were not clustered together. Similar results were obtained for L. andersonii and L. japonicum in Clade 2. The topological structures of the phylogenetic trees inferred using coding genes of chloroplast genomes (Supplementary Figure 2) were different from those of complete cp genome trees. F. arvensis, G. pensylvanica, and G. coarctata formed one calde, which was sister to G. norvegica and G. pensylvanica. Leontopodium species were divided into five main clades; L. forrestianum, L. giraldii, and L. calocephalum formed independent clades, respectively (Supplementary Figure 2). The species in Clades II and V (coding gene tree, Supplementary Figure 2) were identical to those in Clades 2 and 3 (complete genome tree, Figure 5), respectively. The species in Clade 1 (Figure 5) were identical to those in Clades 1, 3, and 4 (Supplementary Figure 2). In addition, the support values of coding genes trees were lower.
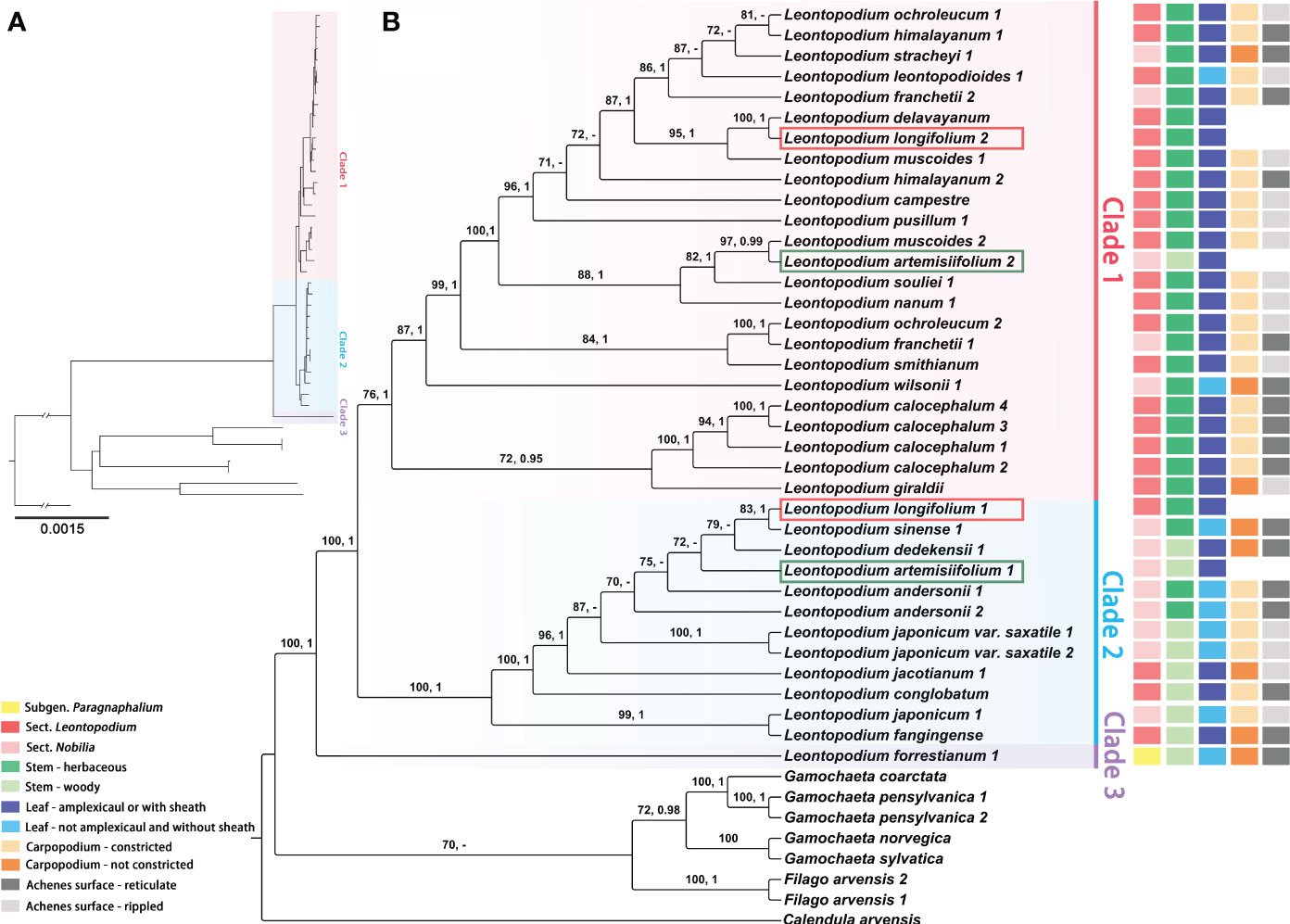
Figure 5 Phylogenetic trees of Leontopodium, Filago, and Gamochaeta, together with Calendula arvensis as an outgroup, were inferred from complete chloroplast genomes. (A) Topology of the ML tree. (B) ML tree with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “–” means that the bootstrap value/posterior probability is less than 70/0.90. The taxonomy system of Leontopodium in Lin (1965) and the morphological characteristics of stems, leaves, and achenes are mapped on the right side.
3.5 Phylogenetic results of nuclear genes
In the phylogenetic trees inferred using ITS, ETS, and the concatenated sequences of ITS and ETS (Figure 6 and Supplementary Figures 3-5), Leontopodium species were clustered into one clade, with strong support (BS = 100; PP = 1). Leontopodium was sister to G. norvegica and G. sylvatica in the phylogenetic trees constructed using ITS and concatenated sequences of ITS and ETS (Figure 6 and Supplementary Figures 3, 5). However, in the ETS phylogenetic tree, Leontopodium was sister to Antennaria taxa (Supplementary Figure 4). Filago species were also clustered into one clade, with strong support, in the phylogenetic trees inferred using nuclear genes; however, the taxa of Antennaria and Gamochaeta were divided into several main clades. For example, in the phylogenetic tree inferred from the concatenated sequences of ITS and ETS (Supplementary Figure 5), Antennaria species were in two main clades (Clades D1 and D2) and Gamochaeta species were in six main clades (Clades B1–B6). Furthermore, six main clades (Clades A1–A6) were formed by the Leontopodium species (Figure 6 and Supplementary Figure 5). Clade A6 was a sister clade to the other clades and included only one species, L. microphyllum. Clade A2, Clade A4, and Clade A5 included 3, 2, and 6 taxa of Leontopodium, respectively. Most Leontopodium taxa were clustered in Clades A1 and A3. The samples of nine taxa, L. artemisiifolium, L. andersonii, L. forrestianum, L. haastioides Hand.-Mazz., L. himalayanum, L. japonicum, L. nanum, L. pusillum and L. souliei, were not clustered together.
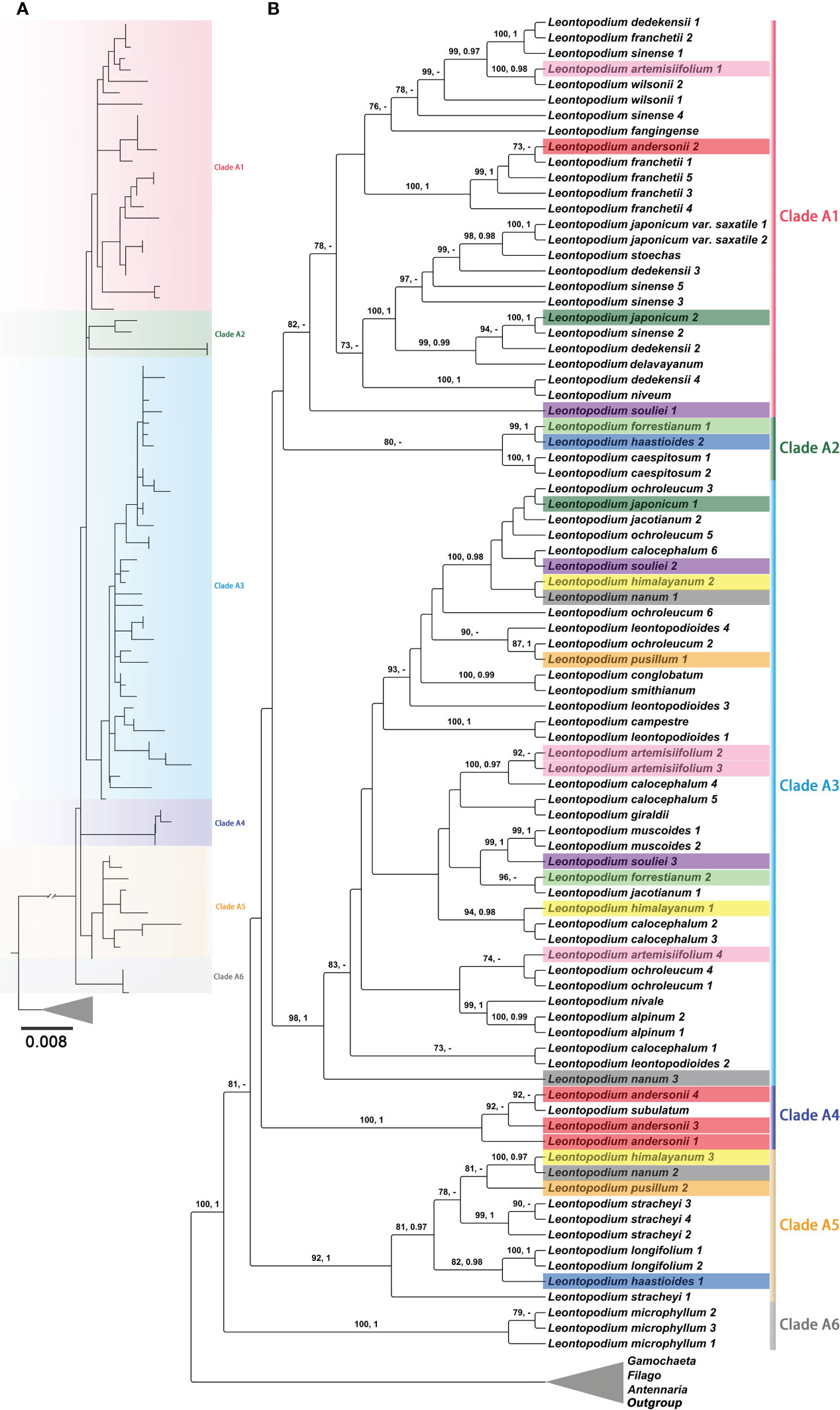
Figure 6 Partial phylogenetic trees of Leontopodium and its closely related genera, together with Calendula arvensis as an outgroup, were inferred from concatenated ITS and ETS sequences. (A) Topology of the ML tree. (B) ML tree, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “–” means that the bootstrap value/posterior probability is less than 70/0.90.
4 Discussion
4.1 Characteristics of cp genomes and genetic variations
In this study, we evaluated 43 complete cp genomes of 30 taxa (one Filago, three Gamochaeta, and 26 Leontopodium), which were assembled de novo for the first time. The plastid genomes of the taxa analyzed here exhibited a typical quadripartite structure, with LSC and SSC regions separated by two IRs, and they were in the genome size range of land plants (Figure 1 and Table 2) (Zheng et al., 2017). In addition, the structure, length, gene number, gene order, and GC content of the plastomes included in our analyses were consistent with those of previously reported Asteraceae cp genomes (Lee et al., 2016; Salih et al., 2017; Ou et al., 2019; Hatmaker et al., 2020; Kim et al., 2020; Loeuille et al., 2021; Pascual-Díaz et al., 2021; Thode et al., 2021; Peng et al., 2022; Yu et al., 2022), which may underscore the overall high stability of cp features at the lower taxonomic level. The cp genomes of Filago, Gamochaeta, and Leontopodium were well conserved in terms of gene number, gene order, and GC content (Table 2). The most remarkable differences among the three genera were the length of the complete cp genome, LSC, SSC, and IR (Table 2). Compared with Filago and Gamochaeta, Leontopodium had a shorter complete cp genome (150,754–151,157 bp), LSC (83,278–83,387 bp), and SSC (17,764–18,092 bp); in contrast, the IR region of Leontopodium was longer than that of Filago and Gamochaeta (Table 2). Furthermore, currently, the NCBI GenBank database contains about 30 cp genomes of Gnaphalieae, and the length of all complete cp genomes of Gnaphalieae reported was longer than 152 kb, such as A. sinica (152,718 bp, KX148081), A. margaritacea var. yedoensis (153,231 bp, LC656264), Helichrysum italicum (Roth) G.Don (152,431 bp, MK089797), Pseudognaphalium oligandrum (DC.) Hilliard & B.L.Burtt (152,797 bp, MK570591), and P. sandwicensium (Gaudich.) Anderb. (152,995 bp, MK570594). Pseudogenization of trnT-GGU was previously reported in Cryptomeria japonica D. Don., and Pelargonium × hortorum (Chumley et al., 2006; Hirao et al., 2008). Lee et al. (2017) determined that the trnT-GGU genes within the cp genomes of A. sinica and L. leiolepis were either pseudogenized or lost and suggested that mutations in the trnT-GGU gene might be used as indicators of generic and/or tribal relationships. In this study, L. artemisiifolium 1 and L. andersonii 1 lacked the trnT-GGU gene (other samples of these two species had the trnT-GGU gene), indicating that the loss of trnT-GGU genes was not associated with species circumscription in Leontopodium. Moreover, Abdullah et al. (2021) performed a broad analysis of the trnT−GGU gene of Asteraceae and found this gene to be a pseudogene in the Asteraceae core, which was linked to an insertion event within the 5′acceptor stem and was not associated with ecological factors such as habit, habitat, and geographical distribution of the species.
In general, the expansion and contraction of IR regions are related to variation in genome length (Raubeson et al., 2007; Wang and Messing, 2011), which is also considered a type of phylogenetic information (Menezes et al., 2018). In the present study, 43 cp genomes were analyzed and the differences between the boundary regions of SC and IR were compared (Figure 2). The genes located at the junctions were well conserved among the 43 cp genomes: rps19 in LSC/IRb, ycf1 in IRb/SSC, ndhF and ψycf1 in SSC/IRa, and ψrps19 and trnH in IRa/LSC. Except for IRb/SSC, the boundaries of LSC/IRb, SSC/Ira, and IRa/LSC were relatively stable. The boundaries of IRb/SSC of Leontopodium were different from those of Filago and Gamochaeta, but were relatively stable at the genus level (Figure 2). Owing to the instability of IRb/SSC, the cp genome experienced IR/SSC contraction and expansion, which may be related to the variation in genome length observed among the three genera. With shifts in the IR/SSC boundaries, IR regions in Leontopodium became longer than those of Filago and Gamochaeta, which suggests that the IR expansion occurred after the splitting of Leontopodium from its sister clade. IRs are thought to stabilize the plastome through homologous recombination-induced repair mechanisms (Wicke et al., 2011). The longer IRs of plastomes are hypothesized to contribute to plastome stabilization because their absence often coincides with severe changes in the gene order (Palmer and Thompson, 1982). However, the cp gene number, length, and order of Leontopodium, Filago, and Gamochaeta were conserved, which indicated that the difference in IR observed among the three genera was insignificant.
Despite the relatively stable and conserved length, structure, gene number, and gene order of the cp genomes, mutation hotspots have been detected. In this study, we used mVISTA to compare the cp genomes of Filago, Gamochaeta, and Leontopodium (Figure 3), and used DnaSP to analyze the percentage of variable loci in the whole cp genomes, 65 coding regions, and 88 non-coding regions (Figure 4 and Supplementary Tables S2, 3). The mVISTA program revealed a relatively high diversity of 43 cp genomes, with genic regions being more conserved than intergenic regions, which is typical of angiosperm cp genomes (Huo et al., 2019; Song et al., 2019; Zhang X. F. et al., 2021). Moreover, as observed in other taxa of Asteraceae (Kim et al., 2020; Loeuille et al., 2021; Pascual-Díaz et al., 2021; Thode et al., 2021; Peng et al., 2022; Yu et al., 2022), the variation in the IR regions was smaller than that observed in the SC regions in the 43 cp genomes analyzed in our study (Figures 3, 4). In addition, the cp genome structure of Leontopodium showed high consistency and was obviously different from, that of Filago and Gamochaeta in some regions, such as matk, trnK (UUU)-rps16, rps16-trnQ (UUG), trnC (GCA)-petN, petN-psbM, and trnE (UUC)-rpoB (Figure 3), which may reflect the phylogenetic relationships among species or genera. In terms of nucleotide diversity, hotspot mutation regions were non-coding, consistent with other cp genomes (Liu et al., 2019; Park et al., 2020; Smidt et al., 2020; Pascual-Díaz et al., 2021). A previous study that investigated Leontopodium phylogeny using cp gene markers, such as matK, trnL, and trnL-trnF, failed to resolve the phylogenetic relationships within the genus (Blöch et al., 2010). Our analyses revealed relatively low nucleotide diversities of matK (Pi = 0.00397), trnL (Pi = 0), and trnL-trnF (Pi = 0.00258) compared to other loci (Figure 4 and Supplementary Tables 2, 3), which explains the low support in the phylogenetic trees that were inferred using these genes (Blöch et al., 2010). Moreover, we detected 14 hotspots (Pi >0.006) in the non-coding regions, which could be used as candidate DNA barcodes for future studies (Supplementary Table 3). Hotspot regions in plants indicate evolution and can be used to distinguish between species or genera (Liu et al., 2019). Therefore, these variable regions may also be useful for assessing the phylogenetic relationships and interspecific differences between Leontopodium species.
4.2 The conflicts of gene trees
The extensive heterogeneity in nucleotide substitution rates among different plastid genes and gene groups is likely to contribute to the phylogenetic ambiguity (Zhang X. et al., 2020; Li et al., 2021). In our phylogenetic analyses, sequence variations differed between the complete cp genome and coding genes datasets (Supplementary Table 4), which led to the different topological structures. This suggests that attention should be paid to the effects of such heterogeneity when functional genes or plastid fragments are used to study the phylogenetic evolution of the Leontopodium cp genome. Guo et al. (2023) indicated that one of the biggest challenges in the field of phylogenomics is the selection of appropriate genomic data for species tree reconstruction. Plastome sequences generally possess slow rates of evolution, in some cases resulting in insufficient numbers of informative sites for resolving rapid radiation (Gitzendanner et al., 2018). The coding genes of chloroplast genomes have fewer informative sites than those of complete chloroplast genomes. Therefore, in our study, the phylogenetic trees inferred using coding genes of chloroplast genomes were more confusing and had lower support values compared with the trees inferred using complete chloroplast genomes, because the non-coding regions of chloroplast genomes could provide more informative sites phylogenetic analysis.
Additionally, topological trees constructed based on complete cp genomes and nrDNA were incongruent (Figures 5, 6 and Supplementary Figures 1, 3-5). Because cp genes and nuclear genes belong to different genetic systems, topological conflicts among different gene trees are a common phenomenon in molecular phylogenetic studies (Zou and Ge, 2008). Frequent hybridization, introgression, horizontal gene transfer, polyploidy accompanied by apomixis, or rapid radiation may have contributed to evolutionary complexity (Zou and Ge, 2008; Liu et al., 2020), and complex phylogenetic relationships are commonly found in Asteraceae (Bayer et al., 2002; Núria et al., 2002; Hidalgo et al., 2006; Kim et al., 2007; Barker et al., 2009; Galbany-Casals et al., 2010; Zhao et al., 2010; Jara-Arancio et al., 2017). The phylogenetic relationships inferred using cp genomes and nrDNA within Leontopodium were complex and incongruent. Leontopodium species were divided into three main clades in the phylogenetic tree obtained from the cp genomes (Figure 5), whereas six main clades were obtained in the phylogenetic tree inferred using nuclear genes (Figure 6). Moreover, two species were clustered into different main clades in the cp tree, whereas nine taxa were clustered in the nrDNA tree (Figures 5, 6). For example, the samples of L. longifolium were divided into different main clades in the cp tree (Figure 5) but were clustered together in the nrDNA tree (Clade A5) (Figure 6). In the phylogenetic tree inferred using nrDNA, the samples of L. longifolium had a close relationship with L. himalayanum, L. nanum, L. pusillum, L. stracheyi, and L. haastioides (Figure 6). In the cp tree, one sample of L. longifolium (L. longifolium 2) was included in Clade 1, together with L. himalayanum, L. nanum, L. pusillum, and L. stracheyi (the cp genome of L. haastioides was not included in our study), whereas the other sample of L. longifolium (L. longifolium 1) was closely related to L. sinense and L. dedekensii (Figure 5). Subsequently, we noticed that three samples (L. longifolium 1, L. sinense 1, and L. dedekensii 1) were all from southern Sichuan, China. This raises the possibility that hybridization occurred between L. longifolium and other species of Leontopodium in Clade 2 (Figure 5), most likely L. sinense or a closely related sympatric species. This caused the capture of a foreign chloroplast haplotype in L. longifolium and fixation of the L. longifolium ITS type. Another example of incongruence resulting from possible hybridization is L. artemisiifolium. The Asteraceae family is highly evolved and is in a stage of rapid differentiation, resulting in a large number of complex and polymorphic transitional taxa in the family, which leads to great difficulties in classification and phylogeny research (Bremer, 1994). The natural hybrids of Leontopodium in China remain controversial (Lin, 1965; Chen et al., 2011), which also indicates complex phylogenetic relationships within this genus. Furthermore, incomplete lineage sorting is a stochastic process that potentially occurs in groups evolving through rapid adaptive radiation. Thus, a phylogeny based on maternally inherited chloroplast genes may not always correspond to a nuclear-gene-based phylogeny (Takahashi et al., 2001; Fior et al., 2013).
4.3 Phylogenetic relationships between Leontopodium and its related genera
All phylogenetic trees inferred using cp genomes and nuclear genes indicated that Leontopodium is monophyletic. In the cp genome phylogenetic tree, Filago and Gamochaeta clustered together and were sister to Leontopodium (Figure 5 and Supplementary Figure 1). In the phylogenetic trees constructed using nrDNA, Filago was monophyletic, but Gamochaeta was nested within Antennaria (Supplementary Figures 3-5). However, Nie et al. (2016) indicated that Antennaria, Leontopodium, and Gamochaeta were monophyletic, whereas Filago was nested with Hesperevax A.Gray, Micropus L., Psilocarphus Nutt., Bombycilaena (DC.) Smoljan., and Evacidium Pomel. The samples included in the phylogenetic analyses can influence the conclusions; for example, A. chilensis J.Rémy and A. linearifolia Wedd. were not included in the study by Nie et al. (2016), but our research included two taxa, which led to the controversy regarding the monophyly of Antennaria. In the ETS tree, Leontopodium was closely related to A. chilensis, A. microphylla Rydb., and A. dioica (L.) Gaertn. (Supplementary Figure 4). Nevertheless, in the phylogenetic trees constructed using ITS and concatenated sequences of nrDNA, Leontopodium is sister to G. norvegica and G. sylvatica (Supplementary Figures 3, 5). Galbany-Casals et al. (2010) indicated that Leontopodium was sister to other genera of the FLAG clade; however, only one species of Leontopodium was analyzed in their study. Three species of Leontopodium and Castroviejoa montelinasana (Em.Schmid) Galbany were clustered into one clade, albeit with low support (Fu et al., 2016). Nie et al. (2016) indicated that Leontopodium was sister to Chionolaena DC., Facelis Cass., Lucilia Cass., Micropsis DC., Stuckertiella Beauverd, Gamochaeta, etc. Thus, the monophyly of Leontopodium is definite, but the relationships between this genus and closely related genera remain unclear. Moreover, more molecular data and taxa should be used to investigate this in the future.
4.4 Phylogenetic relationships within the genus Leontopodium
Compared with nrDNA trees, the support values of cp trees were higher and could also provide clearer infrageneric and interspecies systematic relationships for Leontopodium (Figures 5, 6 and Supplementary Figures 1-5). The relationships within the genus Leontopodium have been previously inferred based on morphological characteristics, such as the morphology and color of the pappus, the shape of bracteal leaves, the indumentum of leaves or achenes, and the types of stems (Handel-Mazzetti, 1928; Lin, 1965). However, our molecular phylogenetic results differed greatly from the morphological classifications. Thus, based on a literature review, natural populations, and specimens of Leontopodium, in the present study, the classifications and phylogenetic relationships of 26 Leontopodium taxa in China were discussed based on the cp phylogenetic tree and the main characteristics of the leaves, stems, and achenes (Figure 5). Both Handel-Mazzetti (1928) and Lin (1965) reported that two subgenera should be included in Leontopodium according to the morphology and color of the pappus, and our results of cp phylogenetic trees also indicated that subgen. Paragnaphalium was sister to subgen. Leontopodium (sect. Nobilia and sect. Leontopodium) (Figure 5). However, in the phylogenetic trees inferred using nrDNA (Figure 6 and Supplementary Figures 3-5), L. forrestianum (subgen. Paragnaphalium) was nested within the subgen. Leontopodium. Therefore, at the subgeneric level, the results of the cp genome phylogenetic analysis were consistent with the classification results obtained from the morphological features of Leontopodium. Lin (1965) indicated the morphology of plant, indumentum, and pappus of L. forrestianum was similar to that of Gnaphalium, but the characters of capitulum and bracteal leave supported this species should be included in Leontopodium, which showed that this species was distantly related to other Leontopodium species. L. forrestianum was the basal taxon of Leontopodium in the phylogenetic tree inferred using the cp genomes (Figure 5). This phenomenon indicated that plastid genomes could better resolve basal phylogenetic relationships because of their conservative property, and particular morphological variation was consistent with the polymorphism of cp genomes.
The two sections (sect. Nobilia and sect. Leontopodium) were included in subgen. Leontopodium, mainly based on plant height, leaves with sheaths or not, and stems being woody or herbaceous (Lin, 1965); however, this division is controversial. First, there were many variations in the height of Leontopodium plants. For example, L. souliei, belonging to sect. Leontopodium was 6–25 cm in height; L. giraldii (sect. Leontopodium) was 10–28 cm in height; L. franchetii belonging to sect. Nobilia was 15–50 cm in height; and L. stracheyi (sect. Nobilia) was 5–60 cm in height (Lin, 1965; Chen et al., 2011). Subsequently, we collated the stem types and found that most species in sect. Leontopodium exhibited herbaceous stems, but a few species had woody stems, such as L. jacotianum, L. conglobatum, and L. fangingense; moreover, most species of sect. Nobilia has woody stems, but a few species also have herbaceous stems, such as L. stracheyi, L. franchetii, L. wilsonii, L. sinense, and L. andersonii (Lin, 1965; Chen et al., 2011). In addition, the morphological characteristics of the leaf bases varied within the sections. The leaves of most of the species in sect. Leontopodium (other than L. leontopodioides and L. microphyllum) were amplexicaul or had sheaths, and the leaves of most species in sect. Nobilia (other than L. stracheyi, L. franchetii, L. artemisiifolium, and L. dedekensii) are not amplexicaul and have no sheaths (Lin, 1965; Chen et al., 2011). The results of our phylogenetic analysis, based on cp genomes, also indicated that sect. Nobilia was nested with sect. Leontopodium, and did not support the monophyly of the two sections (Figure 5). Ma et al. (2022) investigated the achene morphological characteristics of 29 Chinese Leontopodium taxa and divided them into two types based on surface ornamentation (reticulate and rippled). Therefore, we matched the main characteristics (achene surface and carpopodium base) of achene reported by Ma et al. (2022) with the cp genome phylogenetic tree but found that species with the same achene characteristics did not cluster together (Figure 5). It follows that the previous division of the subgen. Leontopodium, based on its morphological characteristics is unreasonable. Although, at the section level, our molecular phylogenetic results were inconsistent with the previous taxonomy system based on morphological characteristics, we found that the characteristics of leaf base, stem types, and carpopodium base had phylogenetic correlation and had potential value in the taxonomic study of Leontopodium. In the phylogenetic trees inferred using chloroplast genomes, the subgen. Leontopodium was divided into two clades (Clades 1 and 2; Figure 5 and Supplementary Figure 1), with most species in Clade 1 having herbaceous stems, amplexicaul or sheathed leaves, and constricted carpopodium; most species in Clade 2 had woody stems, not amplexicaul and sheathed leaves, and not constricted carpopodium (Figure 5 and Supplementary Figure 1). The phylogenetically conserved pattern of leaf base, stem type, and carpopodium base in Leontopodium might be due to greater genetic constraints and/or stabilizing selection pressure favoring stasis of these characters in alpine habitats. Interestingly, similar examples were found in the Anaphalis DC. (Asteraceae, Gnaphalieae), a genus with approximately 110 species, and is also the most diversified in the eastern Himalayas and Hengduan Mountains. A recent phylogenetic analysis of Anaphalis also suggested a higher degree of homoplasy in the leaf base than in other characters (Nie et al., 2013).
The interspecies relationships of Leontopodium are more complex than those of the infrageneric taxonomy system. For example, in the cp genome phylogenetic tree, L. ochroleucum, L. himalayanum, L. stracheyi, L. leontopodioides, and L. franchetii were clustered together with strong support (BS = 86; PP = 1) (Figure 5); however, these five species were dispersed into three main clades in the nrDNA tree (Clades A1, A3, and A5; Figure 6). Moreover, they belong to different sections, subsections, and series, based on their morphological characteristics (Handel-Mazzetti, 1928; Lin, 1965). Although the analysis of complete cp genomes allows the clarification of interspecies relationships, it might be insufficient to fully resolve all phylogenetic relationships, especially in rapidly differentiated Asteraceae (Wortley et al., 2005; Petersen et al., 2011; Li et al., 2015; Hatmaker et al., 2020; Kim et al., 2020; Loeuille et al., 2021; Wang et al., 2021). Because the plastome is regarded as a linked single locus due to its uniparental inheritance, multilocus approaches (including mitochondrial genomes and more nuclear genes) are needed to generate abundant and detailed molecular data for systematic classification and evolutionary research. Thus, future studies should analyze additional specimens from wild populations and obtain more extensive genetic and morphological data to verify the taxonomic and phylogenetic identities of Leontopodium species.
5 Conclusions
In this study, we analyzed the characteristics of 43 cp genomes of Leontopodium and its closely related genera. Subsequently, the phylogenetic position of Leontopodium and the relationships within this genus were inferred based on complete cp genomes and nrDNA. Finally, together with the morphological characteristics, the relationships within Leontopodium were identified and discussed. The cp genomes of Leontopodium, Filago, and Gamochaeta exhibited a typical quadripartite structure, including 85 protein-coding genes, 36–37 tRNA genes, and eight rRNA genes, with a total length of 150,754–151,574 bp. Compared with Filago and Gamochaeta, Leontopodium had longer whole cp genomes and LSC and SSC regions, whereas the length of the IRs was slightly shorter. Genes located at the junctions were well-conserved among the 43 cp genomes. Furthermore, the genic and IR regions were more conserved than the intergenic and SC regions, respectively, which is typical of angiosperm cp genomes. In addition, the cp genome structure of Leontopodium exhibited high consistency and was obviously different from those of Filago and Gamochaeta in some regions, such as matk, trnK (UUU)-rps16, petN-psbM, and trnE (UUC)-rpoB. Moreover, we detected 14 hotspots in non-coding regions that could be used as candidate DNA barcodes. All the phylogenetic trees indicated that Leontopodium was monophyletic. However, topological trees constructed using cp genomes and nrDNA were incongruent. Leontopodium species were divided into three main clades in the cp genome phylogenetic tree and six main clades in the nuclear gene phylogenetic tree. Compared with nrDNA trees, cp trees had higher support values and were more effective for phylogenetic resolution. Except for the subgeneric level, our molecular phylogenetic results were inconsistent with the previous taxonomic system (sections, subsections, and series), which was based on morphological characteristics. Nevertheless, we found that the characteristics of the leaf base, stem types, and carpopodium base were phylogenetically correlated and may have potential value in the taxonomic study of Leontopodium. In the phylogenetic trees inferred using chloroplast genomes, the subgen. Leontopodium was divided into two clades (Clades 1 and 2): most species in Clade 1 had herbaceous stems, amplexicaul or sheathed leaves, and constricted carpopodium; most species in Clade 2 had woody stems, not amplexicaul and sheathed leaves, and not constricted carpopodium.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
S-XZ and X-MX conceived of the study. Q-FZ, X-MX, YL, and S-XZ performed fieldwork. X-MX and Q-FZ conducted the laboratory work and wrote the first draft. X-MX, Q-FZ and J-ZS analyzed data. ZW and Z-LW helped improve language. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (31000097 and 31670188).
Acknowledgments
We are grateful to the PE herbarium (Institute of Botany, Chinese Academy of Sciences, Beijing, China) for providing plant material for sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1163065/full#supplementary-material
Supplementary Figure 1 | BI tree of Leontopodium, Filago, and Gamochaeta, together with Calendula arvensis as an outgroup, was inferred from the complete chloroplast genomes.
Supplementary Figure 2 | Phylogenetic trees of Leontopodium and its closely related genera, together with Calendula arvensis as an outgroup, were inferred from coding genes of chloroplast genomes. (A) Topology of the ML tree. (B) Topology of the BI tree. (C) ML tree, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “-” means that the bootstrap value/posterior probability is less than 70/0.90.
Supplementary Figure 3 | Phylogenetic trees of Leontopodium and its closely related genera, together with Calendula arvensis as an outgroup, were inferred from the ITS sequences. (A) Topology of the ML tree. (B) Topology of the BI tree. (C) ML tree, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “-” means that the bootstrap value/posterior probability is less than 70/0.90.
Supplementary Figure 4 | Phylogenetic trees of Leontopodium and its closely related genera, together with Calendula arvensis as an outgroup, were inferred from the ETS sequences. (A) Topology of the ML tree. (B) Topology of the BI tree. (C) ML tree, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “-” means that the bootstrap value/posterior probability is less than 70/0.90.
Supplementary Figure 5 | Phylogenetic trees of Leontopodium and its closely related genera, together with Calendula arvensis as an outgroup, were inferred from the concatenated sequences of ITS and ETS. (A) Topology of the ML tree. (B) Topology of the BI tree. (C) ML tree, with bootstrap values of ML and posterior probabilities of BI shown at each node. Bootstrap values higher than 70 and posterior probabilities higher than 0.90 are indicated on branches. “-” means that the bootstrap value/posterior probability is less than 70/0.90.
References
Abdullah, Mehmood, F., Heidari, P., Rahim, A., Ahmed, I., Poczai, P. (2021). Pseudogenization of the chloroplast threonine (trnT-GGU) gene in the sunflower family (Asteraceae). Sci. Rep. 11, 21122. doi: 10.1038/s41598-021-00510-4
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Anderberg, A. A. (1989). Phylogeny and reclassification of the tribe Inuleae (Asteraceae). Can. J. Bot. 67, 2277–2296. doi: 10.1139/b89-292
Anderberg, A. A. (1991a). Taxonomy and phylogeny of the tribe Gnaphalieae (Asteraceae). Opera Bot. 104, 1–195.
Anderberg, A. A. (1991b). Taxonomy and phylogeny of the tribe Inuleae (Asteraceae). Plant Syst. Evol. 176, 75–123. doi: 10.1007/BF00937947
Anderberg, A. A. (1991c). Taxonomy and phylogeny of the tribe Plucheae (Asteraceae). Plant Syst. Evol. 176, 145–177. doi: 10.1007/bf00937905
Asanuma, M., Zhu, S., Okura, N., Cai, S. Q., Yoshimatsu, K., Komatsu, K. (2019). Genetic polymorphism of Japanese cultivated Rheum species in the internal transcribed spacer region of nuclear ribosomal DNA. J. Nat. Med. 73, 541–554. doi: 10.1007/s11418-019-01298-4
Barkalov, V. Y., Kozyrenko, M. M. (2014). Phylogenetic relationships of Salix L. subg. Salix species (Salicaceae) according to sequencing data of intergenic spacers of the chloroplast genome and ITS rDNA. Russ. J. Genet. 50, 828–837. doi: 10.1134/S1022795414070035
Barker, N. P., Howis, S., Nordenstam, B., Källersjö, M., Eldenäs, P., Griffioen, C., et al. (2009). Nuclear and chloroplast DNA-based phylogenies of Chrysanthemoides Tourn. ex Medik. (Calenduleae; Asteraceae) reveal extensive incongruence and generic paraphyly, but support the recognition of infraspecific taxa in C. monilifera. S. Afr. J. Bot. 75, 560–572. doi: 10.1016/j.sajb.2009.05.006
Bayer, R. J., Breitwieser, I., Ward, J., Puttock, C. (2007). “Tribe gnaphalieae,” in The families and genera of vascular plants, vol. vol. 8 . Eds. Kadereit, J. W., Jeffrey, C. (Heidelberg: Springer), 246–283. doi: 10.1007/978-3-642-39417-1
Bayer, R. J., Greber, D. G., Bagnall, N. H. (2002). Phylogeny of Australian Gnaphalieae (Asteraceae) based on chloroplast and nuclear sequences, the trnL intron, trnL/trnF intergenic spacer, matK, and ETS. Syst. Bot. 27, 801–814. doi: 10.1043/0363-6445-27.4.801
Bayer, R. J., Starr, J. R. (1998). Tribal phylogeny of the Asteraceae based on two non-coding chloroplast sequences, the trnL intron and trnL/trnF intergenic spacer. Ann. Mo. Bot. Gard. 85, 242–256. doi: 10.2307/2992008
Bentham, G. (1873). “Compositae,” in Genera plantarum, vol. 2. Eds. Bentham, G., Hooker, J. D. (London: Reeve), 163–533. doi: 10.5962/bhl.title.747
Blöch, C., Dickoré, W., Samuel, R., Stuessy, T. (2010). Molecular phylogeny of the Edelweiss (Leontopodium, Asteraceae-Gnaphalieae). Edinb. J. Bot. 67, 235–264. doi: 10.1017/S0960428610000065
Bremer, K. (1994). Asteraceae cladistics & classification (Portland: Timer Press). doi: 10.1111/j.1756-1051.1994.tb00632.x
Cassini, H. (1822). “Inulées (Inuleae),” in Dictionnaire des sciences naturelles, ed. 2, vol. vol 23 . Ed. Cuvier, F. (Paris: Le Normant), 559–582.
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6. doi: 10.1093/gigascience/gix120
Chen, Y. S., Zhu, S. X., Bayer, R. J. (2011). “Gnaphalieae,” in Flora of China, vol. vol.20–21 . Eds. Wu, C. Y., Raven, P. H., Hong, D. Y. (Beijing: Science Press and St. Louis: Missouri Botanical Garden Press), 774–818.
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Dong, W., Liu, J., Yu, J., Wang, L., Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7, e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., Lin, K., Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Eldenäs, P., Källersjö, M., Anderberg, A. A. (1999). Phylogenetic placement and circumscription of tribes Inuleae s. str. and Plucheeae (Asteraceae): evidence from sequences of chloroplast gene ndhF. Mol. Phylogenet. Evol. 13, 50–58. doi: 10.1006/mpev.1999.0635
Fior, S., Li, M., Oxelman, B., Viola, R., Hodges, S. A., Ometto, L., et al. (2013). Spatiotemporal reconstruction of the Aquilegia rapid radiation through next-generation sequencing of rapidly evolving cpDNA regions. New Phytol. 198, 579–592. doi: 10.1111/nph.12163
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Fu, Z. X., Jiao, B. H., Nie, B., Zhang, G. J., Gao, T. G., China Phylogeny Consortium (2016). A comprehensive generic-level phylogeny of the sunflower family: Implications for the systematics of Chinese Asteraceae. J. Syst. Evol. 54, 416–437. doi: 10.1111/jse.12216
Galbany-Casals, M., Andrés-Sánchez, S., Garcia-Jacas, N., Susanna, A., Rico, E., Martínez-Ortega, M. M. (2010). How many of Cassini anagrams should there be ? Molecular systematics and phylogenetic relationships in the Filago group (Asteraceae, Gnaphalieae), with special focus on the genus Filago. Taxon 59, 1671–1689. doi: 10.1002/tax.596003
García, C. C., Basso, A. V., González, A. L., Gonzáles, P., Barboza, G. E. (2018). Unraveling the phylogenetic relationships of Nectouxia (Solanaceae): its position relative to Salpichroa. Plant Syst. Evol. 304, 177–183. doi: 10.1007/s00606-017-1460-5
Gatesy, J., Sloan, D. B., Warren, J. M., Baker, R. H., Simmons, M. P., Springer, M. S. (2019). Partitioned coalescence support reveals biases in species-tree methods and detects gene trees that determine phylogenomic conflicts. Mol. Phylogenet. Evol. 139, 106539. doi: 10.1016/j.ympev.2019.106539
Gitzendanner, M. A., Soltis, P. S., Yi, T. S., Li, D. Z., Soltis, D. E. (2018). “Plastome phylogenetics: 30 years of inferences into plant evolution,” in Advances in Botanical Research, vol. vol. 85 . Eds. Chaw, S. M., Jansen, R. K. (Elsevier: Academic Press), 293–313. doi: 10.1016/bs.abr.2017.11.016
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Guo, C., Luo, Y., Gao, L. M., Yi, T. S., Li, H. T., Yang, J. B., et al. (2023). Phylogenomics and the flowering plant tree of life. J. Integr. Plant Biol. 65, 299–323. doi: 10.1111/jipb.13415
Handel-Mazzetti, H. (1928). Systematische monographie der gattung Leontopodium. Beih. Bot. Centralb. 44, 1–178.
Hashim, A. M., Alatawi, A., Altaf, F. M., Qari, S. H., Elhady, M. E., Osman, G. H., et al. (2021). Phylogenetic relationships and DNA barcoding of nine endangered medicinal plant species endemic to Saint Katherine protectorate. Saudi J. Biolog. Sci. 28, 1919–1930. doi: 10.1016/j.sjbs.2020.12.043
Hatmaker, E. A., Wadl, P. A., Rinehart, T. A., Carroll, J., Lane, T. S., Trigiano, R. N., et al. (2020). Complete chloroplast genome comparisons for Pityopsis (Asteraceae). PloS One 15, e0241391. doi: 10.1371/journal.pone.0241391
Hidalgo, O., Garcia-Jacas, N., Garnatje, T., Susanna, A. (2006). Phylogeny of Rhaponticum (Asteraceae, Cardueae-Centaureinae) and related genera inferred from nuclear and chloroplast DNA sequence data: taxonomic and biogeographic implications. Ann. Bot. 97, 705–714. doi: 10.1093/aob/mcl029
Hirao, T., Watanabe, A., Kurita, M., Kondo, T., Takata, K. (2008). Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: diversifed genomic structure of coniferous species. BMC Plant Biol. 8, 70. doi: 10.1186/1471-2229-8-70
Huang, L. Q., Wu, Y. X. (2006). Study on the antibacterial activity of Leontopodium leontopodioides (Willd.) Beauv. in vitro. J. Tradit. Chin. Vet. Med. 25, 5–7. doi: 10.13823/j.cnki.jtcvm.2006.01.001
Huang, C. H., Zhang, C., Liu, M., Hu, Y., Gao, T., Qi, J., et al. (2016). Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 33, 2820–2835. doi: 10.1093/molbev/msw157
Huo, Y., Gao, L., Liu, B., Yang, Y., Kong, S., Sun, Y., et al. (2019). Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Sci. Rep. 9, 12250. doi: 10.1038/s41598-019-48708-x
Hussain, A., Potter, D., Kim, S., Hayat, M. Q., Bokhari, S. A. I. (2019). Molecular phylogeny of Artemisia (Asteraceae-Anthemideae) with emphasis on undescribed taxa from Gilgit-Baltistan (Pakistan) based on nrDNA (ITS and ETS) and cpDNA (psbA-trnH) sequences. Plant Ecol. Evol. 152, 507–520. doi: 10.5091/plecevo.2019.1583
Jansen, R. K., Kaittanis, C., Saski, C., Lee, S. B., Tomkins, J., Alverson, A. J., et al. (2006). Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol. Biol. 6, 32. doi: 10.1186/1471-2148-6-32
Jansen, R. K., Michaels, H. J., Palmer, J. D. (1991). Phylogeny and character evolution in the Asteraceae based on chloroplast DNA restriction site Mapping. Syst. Bot. 16, 98–115. doi: 10.2307/2418976
Jansen, R. K., Ruhlman, T. A. (2012). “Plastid genomes of seed plants,” in Genomics of Chloroplasts and Mitochondria. Eds. Bock, R., Knoop, V. (Germany, Dordrecht: Springer) 35, 103–126. doi: 10.1007/978-94-007-2920-9_5
Jansen, R. K., Wojciechowski, M. F., Sanniyasi, E., Lee, S., Daniell, H. (2008). Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 48, 1204–1217. doi: 10.1016/j.ympev.2008.06.013
Jara-Arancio, P., Vidal, P. M., Panero, J. L., Marticorena, A., Arancio, G., Arroyo, M. T. K. (2017). Phylogenetic reconstruction of the South American genus Leucheria Lag. (Asteraceae, Nassauvieae) based on nuclear and chloroplast DNA sequences. Plant Syst. Evol. 303, 221–232. doi: 10.1007/s00606-016-1366-7
Jiang, W., Tan, W., Gao, H., Yu, X., Zhang, H., Bian, Y., et al. (2020). Transcriptome and complete chloroplast genome of Glycyrrhiza inflata and comparative analyses with the other two licorice species. Genomics 112, 4179–4188. doi: 10.1016/j.ygeno.2020.07.007
Jiao, S. P., Chen, B., Jiang, H., Zhang, L. M. (1997). Studies on the hypoglycemic effect of common edelweiss (Leontopodium leontopodioides). Chin. Tradit. Herb. Drugs 11, 673–675. doi: 10.7501/j.issn.0253-2670.1997.11.1997011350
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., de Pamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Karis, P. O. (1993). Morphological phylogenetics of the Asteraceae-Asteroideae, with notes on character evolution. Plant Syst. Evol. 186, 69–93. doi: 10.1007/BF00937714
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinf. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kim, S., Chunghee, L., Mejías, J. A. (2007). Phylogenetic analysis of chloroplast DNA matK gene and ITS of nrDNA sequences reveals polyphyly of the genus Sonchus and new relationships among the subtribe Sonchinae (Asteraceae: Cichorieae). Mol. Phylogenet. Evol. 44, 578–597. doi: 10.1016/j.ympev.2007.03.014
Kim, G. B., Lim, C. E., Kim, J. S., Kim, K., Lee, J. H., Yu, H. J., et al. (2020). Comparative chloroplast genome analysis of Artemisia (Asteraceae) in East Asia: insights into evolutionary divergence and phylogenomic implications. BMC Genomics 21, 415. doi: 10.1186/s12864-020-06812-7
Kuzmanović, N., Lakušić, D., Frajman, B., Alegro, A., Schönswetter, P. (2017). Phylogenetic relationships in Seslerieae (Poaceae) including resurrection of Psilathera and Sesleriella, two monotypic genera endemic to the Alps. Taxon 66, 1349–1370. doi: 10.12705/666.5
Lee, D. H., Cho, W. B., Choi, B. H., Lee, J. H. (2017). Characterization of two complete chloroplast genomes in the Tribe Gnaphalieae (Asteraceae): gene loss or pseudogenization of trn T-GGU and implications for phylogenetic relationships. Korean J. Hortic. Sci. Technol. 35, 769–783. doi: 10.12972/KJHST.20170081
Lee, Y. S., Park, J. Y., Kim, J. K., Lee, H. O., Park, H. S., Lee, S. C., et al. (2016). The complete chloroplast genome sequences of Artemisia gmelinii and Artemisia capillaris (Asteraceae). Mitochondrial DNA Part B 1, 410–411. doi: 10.1080/23802359.2016.1176880
Leister, D. (2003). Chloroplast research in the genomic age. Trends Genet. 19, 47–56. doi: 10.1016/s0168-9525(02)00003-3
Li, D. Z., Gao, L., Li, H., Wang, H., Ge, X. J., Liu, J., et al. (2011). Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. PNAS 108, 19641–19646. doi: 10.1073/pnas.1104551108
Li, L., Hu, Y., He, M., Zhang, B., Wu, W., Cai, P., et al. (2021). Comparative chloroplast genomes: insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genomics 22, 138. doi: 10.1186/s12864-021-07427-2
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., Chen, S. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. Cambridge Philos. Soc 90, 157–166. doi: 10.1111/brv.12104
Lin, Y. (1965). Notulae de nonnulis generibus tribus Inulearum familiae Compositarum florae sinicae. Acta Phytotaxon. Sin. 10, 167–181.
Lin, Y. (1979). “Leontopodium R. Br. ex Cass,” in Flora of China, vol. vol 75 . Ed. The editorial committee of Flora of China, Chinese academy of sciences (Beijing: Science Press), 72–141.
Liu, B. B., Campbell, C. S., Hong, D. Y., Wen, J. (2020). Phylogenetic relationships and chloroplast capture in the Amelanchier-Malacomeles-Peraphyllum clade (Maleae, Rosaceae): Evidence from chloroplast genome and nuclear ribosomal DNA data using genome skimming. Mol. Phylogenet. Evol. 147, 106784. doi: 10.1016/j.ympev.2020.106784
Liu, Y., Huo, N., Dong, L., Wang, Y., Zhang, S., Young, H. A., et al. (2013). Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PloS One 2, e57533. doi: 10.1371/journal.pone.0057533
Liu, E., Yang, C., Liu, J., Jin, S., Harijati, N., Hu, Z., et al. (2019). Comparative analysis of complete chloroplast genome sequences of four major Amorphophallus species. Sci. Rep. 9, 809. doi: 10.1038/s41598-018-37456-z
Loeuille, B., Thode, V., Siniscalchi, C., Andrade, S., Rossi, M., Pirani, J. R. (2021). Extremely low nucleotide diversity among thirty-six new chloroplast genome sequences from Aldama (Heliantheae, Asteraceae) and comparative chloroplast genomics analyses with closely related genera. PeerJ 9, e10886. doi: 10.7717/peerj.10886
Lowe, T. M., Chan, P. P. (2016). TRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, 54–57. doi: 10.1093/nar/gkw413
Ma, Y., Wei, Z., Lu, Y., Li, Z., Zhao, Q., Zhu, S. (2022). Achene micromorphological characteristics and taxonomic significance in Leontopodium R. Brown ex Cass. taxa (Asteraceae: Gnaphalieae) in China. Pak. J. Bot. 54, 1461–1474. doi: 10.30848/PJB2022-4
Malekmohammadi, M., Akhani, H., Borsch, T. (2017). Phylogenetic relationships of Limonium (Plumbaginaceae) inferred from multiple chloroplast and nuclear loci. Taxon 66, 1128–1146. doi: 10.12705/665.8
Mandel, J. R., Dikow, R. B., Siniscalchi, C. M., Thapa, R., Watson, L. E., Funk, V. A. (2019). A fully resolved backbone phylogeny reveals numerous dispersals and explosive diversifications throughout the history of Asteraceae. PNAS 116, 14083–14088. doi: 10.1073/pnas.1903871116
Menezes, A. P. A., Resende-Moreira, L. C., Buzatti, R. S. O., Nazareno, A. G., Carlsen, M., Lobo, F. P., et al. (2018). Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci. Rep. 8, 2210. doi: 10.1038/s41598-018-20189-4
Merxmüller, H., Leins, P., Roessler, H. (1977). “Inuleae: systematic review,” in The biology and chemistry of the Compositae. Eds. Heywood, V. H., Harborne, J. B., Tumer, B. L. (London: Academic Press), 577–602. doi: 10.2307/2418758
Ng, P. K., Lin, S. M., Lim, P. E., Liu, L. C., Pai, T. W. (2017). Complete chloroplast genome of Gracilaria firma (Gracilariaceae, Rhodophyta), with discussion on the use of chloroplast phylogenomics in the subclass Rhodymeniophycidae. BMC Genomics 18, 40. doi: 10.1186/s12864-016-3453-0
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, Z. L., Funk, V. A., Meng, Y., Deng, T., Sun, H., Wen, J. (2016). Recent assembly of the global herbaceous flora: evidence from the paper daisies (Asteraceae: Gnaphalieae). New Phytol. 209, 1795–1806. doi: 10.1111/nph.13740
Nie, Z. L., Funk, V. A., Sun, H., Deng, T., Meng, Y., Wen, J. (2013). Molecular phylogeny of Anaphalis (Asteraceae, Gnaphalieae) with biogeographic implications in the Northern Hemisphere. J. Plant Res. 126, 17–32. doi: 10.1007/s10265-012-0506-6
Núria, G. J., Teresa, G., Alfonso, S., Roser, V. (2002). Tribal and subtribal delimitation and phylogeny of the Cardueae (Asteraceae): A combined nuclear and chloroplast DNA analysis. Mol. Phylogenet. Evol. 22, 51–64. doi: 10.1006/mpev.2001.1038
Ou, C. Z., Feng, Y. L., Hu, Y. K., Tian, X. Y., Fu, Z. X. (2019). Characterization of the complete chloroplast genome sequence of Aster hersileoides (Asteraceae, Astereae) and its phylogenetic implications. Mitochondrial DNA Part B 4, 985–986. doi: 10.1080/23802359.2019.1581109
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.ge.19.120185.001545
Palmer, J. D., Thompson, W. F. (1982). Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 29, 537–550. doi: 10.1016/0092-8674(82)90170-2
Panero, J. L., Crozier, B. S. (2016). Macroevolutionary dynamics in the early diversification of Asteraceae. Mol. Phylogenet. Evol. 99, 116–132. doi: 10.1016/j.ympev.2016.03.007
Park, I., Yang, S., Song, J. H., Moon, B. C. (2020). Dissection for floral micromorphology and plastid genome of valuable medicinal Borages Arnebia and Lithospermum (Boraginaceae). Front. Plant Sci. 11. doi: 10.3389/fpls.2020.606463
Pascual-Díaz, J. P., Garcia, S., Vitales, D. (2021). Plastome diversity and phylogenomic relationships in Asteraceae. Plants 10, 2699. doi: 10.3390/plants10122699
Peng, J. Y., Zhang, X. S., Zhang, D. G., Wang, Y., Deng, T., Huang, X. H., et al. (2022). Newly reported chloroplast genome of Sinosenecio albonervius Y. Liu & Q. E. Yang and comparative analyses with other Sinosenecio species. BMC Genomics 23, 639. doi: 10.1186/s12864-022-08872-3
Petersen, G., Aagesen, L., Seberg, O., Larsen, I. H. (2011). When is enough, enough in phylogenetics? A case in point from Hordeum (Poaceae). Cladistics 27, 428–446. doi: 10.1111/j.1096-0031.2011.00347.x
Pirani, A., Moazzeni, H., Zarre, S., Rabeler, R. K., Oxelman, B., Pavlenko, A. V., et al. (2020). Phylogeny of Acanthophyllum s.l. revisited: an update on generic concept and sectional classification. Taxon 69, 500–514. . doi: 10.1002/tax.12241
Qian, S., Zhang, Y., Lee, S. Y. (2021). Comparative analysis of complete chloroplast genome sequences in Edgeworthia (Thymelaeaceae) and new insights into phylogenetic relationships. Front. Genet. 12. doi: 10.3389/fgene.2021.643552
Rambaut, A. (2014) Molecular Evolution, Phylogenetics and Epidemiology. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (Accessed May 31, 2019).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8, 174. doi: 10.1186/1471-2164-8-174
Rogalski, M., do Nascimento, V. L., Fraga, H. P., Guerra, M. P. (2015). Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00586
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Ross, T. G., Barrett, C. F., Gomez, M. S., Lam, V. K. Y., Henriquez, C. L., Les, D. H., et al. (2016). Plastid phylogenomics and molecular evolution of Alismatales. Cladistics 32, 160–178. doi: 10.1111/cla.12133
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Safer, S., Tremetsberger, K., Guo, Y. P., Kohl, G., Samuel, M. R., Stuessy, T. F., et al. (2011). Phylogenetic relationships in the genus Leontopodium (Asteraceae: Gnaphalieae) based on AFLP data. Bot. J. Linn. Soc 165, 364–377. doi: 10.1111/j.1095-8339.2011.01117.x
Salih, R. H. M., Majeský, L., Schwarzacher, T., Gornall, R., Heslop-Harrison, P. (2017). Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): Identity and variation between three microspecies. PloS One 12, e0168008. doi: 10.1371/journal.pone.0168008
Sam, B. L., Wendy, L. C., Finn, K., Nina, R. (2017). First plastid phylogenomic study reveals potential cyto-nuclear discordance in the evolutionary history of Ficus L. (Moraceae). Mol. Phylogenet. Evol. 109, 93–104. doi: 10.1016/j.ympev.2016.12.031
Smidt, E. C., Páez, M. Z., Vieira, L., Viruel, J., de Baura, V. A., Balsanelli, E., et al. (2020). Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PloS One 15, e0227991. doi: 10.1371/journal.pone.0227991
Smissen, R. D., Bayer, R. J., Bergh, N. G., Breitwieser, I., Freire, S. E., Galbany-Casals, M., et al. (2020). A revised subtribal classification of Gnaphalieae (Asteraceae). Taxon 69, 778–806. doi: 10.1002/tax.12294
Song, Y., Zhang, Y., Xu, J., Li, W., Li, M. (2019). Characterization of the complete chloroplast genome sequence of Dalbergia species and its phylogenetic implications. Sci. Rep. 9, 20401. doi: 10.1038/s41598-019-56727-x
Stöver, B. C., Müller, K. F. (2010). TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf. 11, 7. doi: 10.1186/1471-2105-11-7
Sun, J., Sun, R., Liu, H., Chang, L., Li, S., Zhao, M., et al. (2021). Complete chloroplast genome sequencing of ten wild Fragaria species in China provides evidence for phylogenetic evolution of Fragaria. Genomics 113, 1170–1179. doi: 10.1016/j.ygeno.2021.01.027
Takahashi, K., Terai, Y., Nishida, M., Okada, N. (2001). Phylogenetic relationships and ancient incomplete lineage sorting among cichlid fishes in Lake Tanganyika as revealed by analysis of the insertion of retroposons. Mol. Biol. Evol. 18, 2057–2066. doi: 10.1093/oxfordjournals.molbev.a003747
Tan, W. H., Chai, L. C., Chin, C. F. (2020). Efficacy of DNA barcode internal transcribed spacer 2 (ITS2) in phylogenetic study of Alpinia species from Peninsular Malaysia. Physiol. Mol. Biol. Plants 26, 1889–1896. doi: 10.1007/s12298-020-00868-1
Thode, V. A., Oliveira, C. T., Loeuille, B., Siniscalchi, C. M., Pirani, J. R. (2021). Comparative analyses of Mikania (Asteraceae: Eupatorieae) plastomes and impact of data partitioning and inference methods on phylogenetic relationships. Sci. Rep. 11, 13267. doi: 10.1038/s41598-021-92727-6
Tian, S., Lu, P., Zhang, Z., Wu, J. Q., Zhang, H., Shen, H. (2021). Chloroplast genome sequence of Chongming lima bean (Phaseolus lunatus L.) and comparative analyses with other legume chloroplast genomes. BMC Genomics 22, 194. doi: 10.1186/s12864-021-07467-8
Twyford, A. D., Ness, R. W. (2017). Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 17, 858–868. doi: 10.1111/1755-0998.12626
Vicent, M., Galán, J. M. G., Sessa, B. (2017). Phylogenetics and historical biogeography of Lomaridium (Blechnaceae: Polypodiopsida). Taxon 66, 1304–1316. doi: 10.12705/666.3
Wang, W., Messing, J. (2011). High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PloS One 6, e24670. doi: 10.1371/journal.pone.0024670
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., Guo, L. (2021). Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genomic 22, 103. doi: 10.1186/s12864-021-07394-8
Wei, R., Yan, Y. H., Harris, A. J., Kang, J. S., Shen, H., Xiang, Q. P., et al. (2017). Plastid phylogenomics resolve deep relationships among Eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol. Evol. 9, 1646–1657. doi: 10.1093/gbe/evx107
Wicke, S., Schneeweiss, G. M., de Pamphilis, C. W., Müller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wortley, A. H., Rudall, P. J., Harris, D. J., Scotland, R. W. (2005). How much data are needed to resolve a difficult phylogeny ? Case study in Lamiales. Syst. Biol. 54, 697–709. doi: 10.1080/10635150500221028
Wu, C. S., Wang, T. J., Wu, C. W., Wang, Y. N., Chaw, S. M. (2017). Plastome evolution in the sole hemiparasitic genus laurel dodder (Cassytha) and insights into the plastid phylogenomics of Lauraceae. Genome Biol. Evol. 9, 2604–2614. doi: 10.1093/gbe/evx177
Wu, N. Z., Zhan, R., Gou, P. (2013). Anti-oxidation active components and structure analysis from Leontopodium leontopodioides. Nat. Prod. Res. Dev. 25, 296–301. doi: 10.16333/j.1001-6880.2013.03.002
Xiong, A. S., Peng, R. H., Zhuang, J., Gao, F., Zhu, B., Fu, X. Y., et al. (2009). Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnol. Adv. 27, 340–347. doi: 10.1016/j.biotechadv.2009.01.012
Yang, H. M., Zhang, Y. X., Yang, J. B., Li, D. Z. (2013). The monophyly of Chimonocalamus and conflicting gene trees in Arundinarieae (Poaceae: Bambusoideae) inferred from four plastid and two nuclear markers. Mol. Phylogenet. Evol. 68, 340–356. doi: 10.1016/j.ympev.2013.04.002
Yang, Y., Zhu, J., Feng, L., Zhou, T., Bai, G., Yang, J., et al. (2018). Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00082
Yu, S., Yang, X., Tian, X., Liu, X., Lu, X., Huang, C., et al. (2022). The complete chloroplast genome sequence of the monotypic and enigmatic genus Cavea (tribe Gymnarrheneae) and a comparison with other species in Asteraceae. J. Genet. 101, 20. doi: 10.1007/s12041-022-01360-3
Zhang, M. Y., Fritsch, P. W., Ma, P. F., Wang, H., Lu, L., Li, D. Z. (2017). Plastid phylogenomics and adaptive evolution of Gaultheria series Trichophyllae (Ericaceae), a clade from sky islands of the Himalaya-Hengduan Mountains. Mol. Phylogenet. Evol. 110, 7–18. doi: 10.1016/j.ympev.2017.01.015
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, C., Huang, C. H., Liu, M., Hu, Y., Panero, J. L., Luebert, F., et al. (2021). Phylotranscriptomic insights into Asteraceae diversity, polyploidy, and morphological innovation. J. Integr. Plant Biol. 63, 1273–1293. doi: 10.1111/jipb.13078
Zhang, X. F., Landis, J. B., Wang, H. X., Zhu, Z. X., Wang, H. F. (2021). Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 21, 219. doi: 10.1186/s12870-021-02985-9
Zhang, X., Sun, Y., Landis, J. B., Lv, Z., Shen, J., Zhang, H., et al. (2020). Plastome phylogenomic study of Gentianeae (Gentianaceae): widespread gene tree discordance and its association with evolutionary rate heterogeneity of plastid genes. BMC Plant Biol. 20, 340. doi: 10.1186/s12870-020-02518-w
Zhao, H. B., Chen, F. D., Chen, S. M., Wu, G. S., Guo, W. M. (2010). Molecular phylogeny of Chrysanthemum, Ajania and its allies (Anthemideae, Asteraceae) as inferred from nuclear ribosomal ITS and chloroplast trnL-F IGS sequences. Plant Syst. Evol. 284, 153–169. doi: 10.1007/s00606-009-0242-0
Zheng, X. M., Wang, J., Feng, L., Liu, S., Pang, H., Qi, L., et al. (2017). Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 7, 1555. doi: 10.1038/s41598-017-01518-5
Zhou, J., Gao, Y. Z., Wei, J., Liu, Z. W., Downie, S. R. (2020). Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA its sequences: Rampant polyphyly, placement of the Chinese endemic species, and a much-reduced circumscription of the genus. Int. J. Plant Sci. 181, 306–323. doi: 10.1086/706851
Zhou, X. M., Zhang, L. B. (2017). Nuclear and plastid phylogenies suggest ancient intersubgeneric hybridization in the fern genus Pyrrosia (Polypodiaceae), with a classification of Pyrrosia based on molecular and non-molecular evidence. Taxon 66, 1065–1048. doi: 10.12705/665.5
Keywords: Leontopodium, chloroplast genome, genome structure, nuclear ribosomal DNA, phylogenetic analysis
Citation: Xu X-M, Wei Z, Sun J-Z, Zhao Q-F, Lu Y, Wang Z-L and Zhu S-X (2023) Phylogeny of Leontopodium (Asteraceae) in China—with a reference to plastid genome and nuclear ribosomal DNA. Front. Plant Sci. 14:1163065. doi: 10.3389/fpls.2023.1163065
Received: 10 February 2023; Accepted: 10 July 2023;
Published: 31 July 2023.
Edited by:
Daniel Pinero, National Autonomous University of Mexico, MexicoReviewed by:
Zelong Nie, Jishou University, ChinaSeonJoo Park, Yeungnam University, Republic of Korea
Copyright © 2023 Xu, Wei, Sun, Zhao, Lu, Wang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shi-Xin Zhu, sxzhu@zzu.edu.cn