 Ke-Wang Xu1†
Ke-Wang Xu1† Xue-Fen Wei2†
Xue-Fen Wei2† Chen-Xue Lin1Min Zhang1,3Qiang Zhang1Peng Zhou3
Chen-Xue Lin1Min Zhang1,3Qiang Zhang1Peng Zhou3 Yan-Ming Fang1
Yan-Ming Fang1 Jia-Yu Xue2*Yi-Fan Duan1*
Jia-Yu Xue2*Yi-Fan Duan1*- 1Key Laboratory of National Forestry and Grassland Administration on Subtropical Forest Biodiversity Conservation, Co-innovation Center for Sustainable Forestry in Southern China, College of Biology and the Environment, Nanjing Forestry University, Nanjing, China
- 2College of Horticulture, Academy for Advanced Interdisciplinary Studies, Nanjing Agricultural University, Nanjing, China
- 3Jiangsu Academy of Forestry, Nanjing, China
The Ilex L. (hollies) genus of Aquifoliaceae shows high species diversity in tropical and subtropical regions of Asia and South America. Throughout the range of the genus, Ilex species have been widely used in beverage and medicine production and as ornamentals. Here, we assembled a high-quality, chromosome-level genome of Ilex latifolia, which has extremely high economic value because of its useful secondary metabolite production and the high ornamental value of its decorative red berries. The 99.8% genome sequence was anchored to 20 pseudochromosomes, with a total length of 766.02 Mb and a scaffold N50 of 33.45 Mb. Based on the comparative genomic analysis of 14 angiosperm species, we recovered I. latifolia as the sister group to all other campanulids. Two whole-genome duplication (WGD) events were identified in hollies: one shared ancient WGD in the ancestor of all eudicots and a recent and independent WGD in hollies. We performed a genome-wide search to screen candidate genes involved in the biosynthesis of pentacyclic triterpenoid saponins in I. latifolia. Three subfamilies of CYP450 (CYP71A, CYP72A, and CYP716A) appear to have expanded. The transcriptomic analysis of I. latifolia leaves at five developmental stages revealed that two CYP716A genes and one CYP72A gene probably play important roles in this biosynthetic pathway. In addition, we totally identified 12 genes in the biosynthesis pathways of pelargonidin and cyanidin and observed their differential expression in green and red fruit pericarps, suggesting an association between pelargonidin and cyanidin biosynthesis and fruit pericarp color change. The accumulation of pelargonidin and cyanidin is expected to play an important role in the ornamental value of I. latifolia. Altogether, this study elucidated the molecular basis of the medicinal and ornamental value of I. latifolia, providing a data basis and promising clues for further applications.
Introduction
Ilex L. (the hollies) is a genus of trees, shrubs, and (rarely) climbers within the Aquifoliaceae family that contains more than 600 species with an irregular cosmopolitan distribution, in which most species occur in tropical and subtropical regions of South America and Asia (Loizeau et al., 2016). Hollies have high economic and ornamental value. The leaves of over 60 species of this genus are used in beverages (Loizeau et al., 2016) such as Paraguay tea (mate tea) made from Ilex paraguariensis A. St.-Hil., which is drunk throughout South America (Bracesco et al., 2011), and the black drink, or “Cassena,” made from Ilex vomitoria Aiton, which has been used by certain native North Americans (Folch, 2021). Many Ilex species (e.g., American holly, Ilex opaca Aiton; Japanese holly, Ilex crenata Thunb.; and Ilex purpure Hassk. in China) are widely grown in parks and gardens throughout the world for their foliage and decorative berries (e.g., on Christmas trees) and hold an important position in gardens worldwide. However, genetic research on holly species has mainly focused on evaluating their genetic diversity by identifying molecular markers, determining phylogenetic relationships, and revealing speciation and lineage diversification until now (Gottlieb et al., 2005; Selbach-Schnadelbach et al., 2009; Manen et al., 2010; Shi et al., 2016; Xu et al., 2021; Yao et al., 2021). The weak foundation of genome-wide research not only for the entire family Aquifoliaceae but also for the order Aquifoliales is not equal to the importance of these groups.
Ilex latifolia Thunb. is a subtropical evergreen tree native to China and Japan (Figure 1A). In addition to its ornamental functions, the tender leaves of this plant can be processed into a specific kind of tea known as Kudingcha (Sun et al., 2011). Kudingcha has a slight bitter taste, and its Chinese name clearly reflects its bitter flavor; it is known as a very healthy drink and is a traditional Chinese medicine with a long history in southern China because the young leaves of I. latifolia contain pentacyclic triterpenoid saponins and flavonoids, which have blood lipid- and blood pressure-lowering, detoxification and cancer-combating effects (Li et al., 2013; Yang et al., 2015). Because of these benefits to human health, Kudingcha has become the plant-based beverage with the highest production in China, second only to tea [from young leaves of Camellia sinensis (L.) Kuntze]. However, the genome of I. latifolia has not been sequenced, and candidate genes associated with some important active ingredients of Kudingcha and the ornamental traits of the genus have yet to be identified. The lack of transcriptional and genomic information on I. latifolia has greatly limited genetic and breeding research on this species and its closely related congeners in the genus. Therefore, more detailed molecular and genomic resources are still needed to investigate the genomic signatures of holly species.
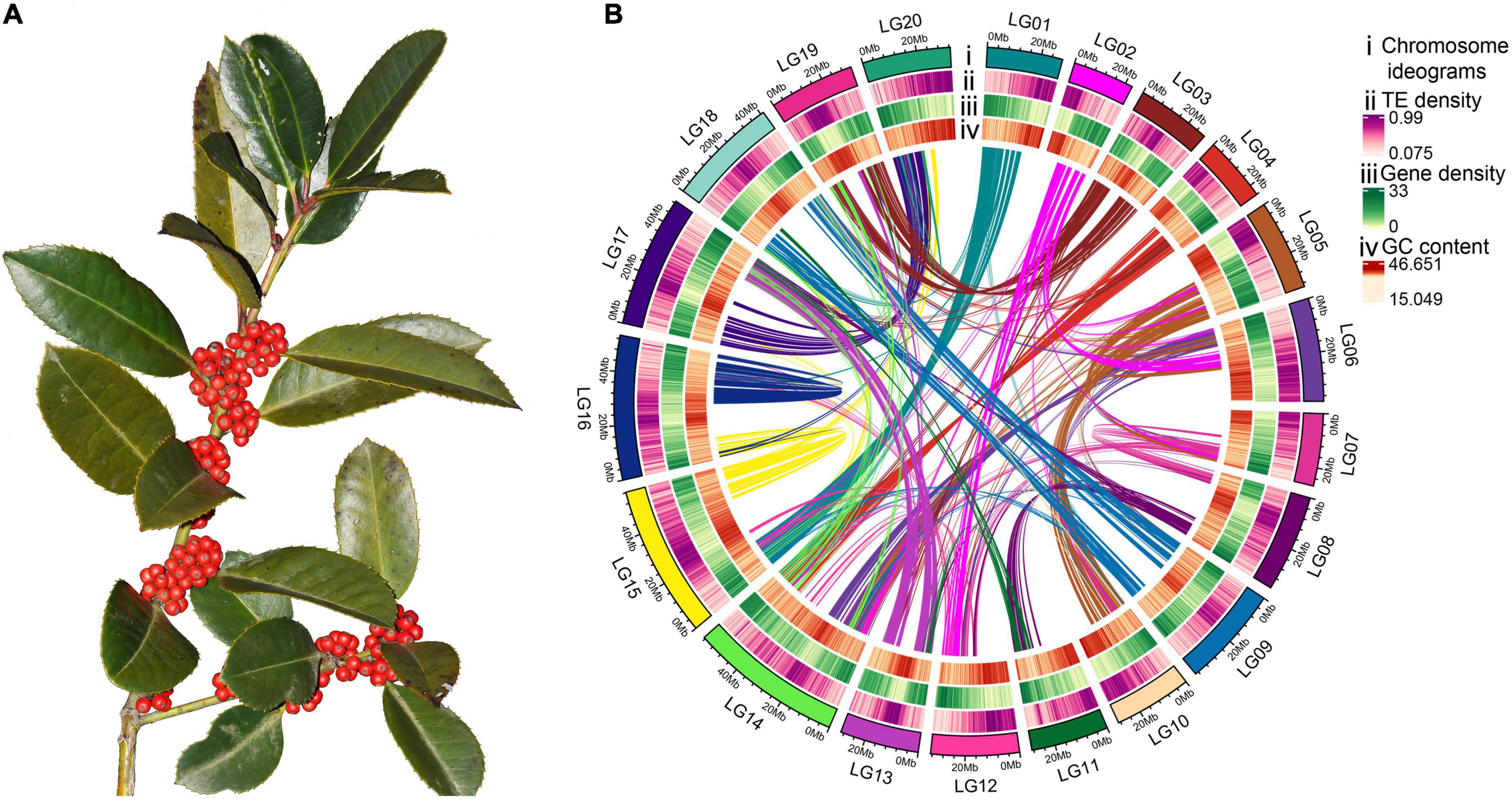
Figure 1. Morphology and high-quality genome assembly of Ilex latifolia. (A) Fruiting branches of I. latifolia showing its ornamental berries. (B) The genome features across 20 chromosomes of I. latifolia. From outermost to innermost, the circles show chromosome ideograms; TE, transposable element (long and short interspersed nuclear elements) density; gene density; GC content and colinear blocks in the genome.
Recently, Yao et al. (2022) assembled the first holly genome of a deciduous species [Ilex polyneura (Hand.-Mazz.) S.Y. Hu] and performed a population genomics study to clarify biogeographic ambiguities but did not focus on the molecular mechanisms underlying the medicinal and ornamental value of hollies. In this study, we therefore sequenced and assembled a chromosome-level genome for I. latifolia using Nanopore technology combined with Hi-C scaffolding and analyzed its evolutionary and genomic features. In addition, we performed a transcriptomic analysis of its leaves at multiple developmental stages to identify critical genes involved in the biosynthesis of pentacyclic triterpenoid saponins. Candidate genes associated with fruit anthocyanin biosynthesis were also identified via a combined analyses of transcriptomic data at two different developmental stages. This study provides the second comprehensive Ilex genome reported to date and provides insight into the biosynthesis of pentacyclic triterpenoid saponins and the coloration of fruits in economic and ornamental tree plants.
Materials and methods
Sample collection and Illumina and Nanopore sequencing
In this study, all materials used for genome sequencing were collected from an adult I. latifolia plant growing on the campus of Nanjing Forestry University. Approximately 500 mg of tissue was dissected and stored in liquid nitrogen until it was delivered in dry ice. Total genomic DNA extraction was performed by using a sodium dodecyl sulfate (SDS)-based method before purification with chloroform for Illumina and Nanopore sequencing. For Illumina sequencing, DNA was sonicated to a fragment size of 500 bp with an ultrasonicator, and the library was then prepared by using an NEB Ultra DNA library prep kit (NEB, United Kingdom) according to the manufacturer’s instructions. The paired-end sequencing of the libraries was performed on a NovaSeq 6000 system. A total of 88.62 Gb of raw data were obtained, and unpaired reads, low-quality reads, connector contamination, and duplicated reads were filtered to obtain clean data (Supplementary Table 1).
For Nanopore sequencing, 2 μg of gDNA was repaired using the NEB Next FFPE DNA Repair Mix kit (M6630, United States) and subsequently processed using the ONT Template prep kit (SQK-LSK109, United Kingdom) according to the manufacturer’s instructions. The large-segment library was premixed with loading beads and then pipetted into a previously used and washed R9 flow cell. The library was sequenced on the ONT PromethION platform with the corresponding R9 cell and ONT sequencing reagent kit (EXP-FLP001.PRO.6, United Kingdom) according to the manufacturer’s instructions.
Estimation of genome size, heterozygosity, and repeat content
Before genome assembly, the read information obtained by sequencing was subjected to K-mer analysis. The occurrence of k-mers was counted with Jellyfish (Marçais and Kingsford, 2011). The general features of the genome, including its repeat contents, heterozygosity rates, and genome size, were estimated with GenomeScope (Vurture et al., 2017).
De novo genome assembly of Nanopore reads and assembly assessment
Nanopore reads were initially corrected using Canu (Koren et al., 2017) and then used as input data for SMARTdenovo1 assembly. After completing the initial assembly, Racon (Vaser et al., 2017) and Pilon (Walker et al., 2014) were used to calibrate and polish the assembled reference genome by using Nanopore and Illumina data. In addition, CEGMA v2.5 (Parra et al., 2007) and BUSCO v2.0 (Simão et al., 2015) were used to assess the genome completeness and gene set completeness of the draft genome sequences (Supplementary Tables 2, 3).
Hi-C chromosome assembly
The adapter sequences of the raw Hi-C reads were trimmed, and low-quality paired-end reads were removed to obtain clean data. Then, the clean reads were aligned to the assembled results using BWA v0.7.10-r789 (Li and Durbin, 2009). Invalid read pairs, including dangling-end, self-circularized, religated and dumped products, were filtered, and valid interaction read pairs were identified and retained from the uniquely mapped paired-end reads with HiC-Pro v2.8.12 (Servant et al., 2015). These corrected scaffolds were then clustered, ordered, and oriented onto chromosomes using LACHESIS (Belton et al., 2012) with the following parameters: CLUSTER_MIN_RE_SITES = 5, CLUSTER_MAX_LINK_DENSITY = 2, CLUSTER NONINFORMATIVE RATIO = 2, ORDER MIN N RES IN TRUNK = 5, ORDER MIN N RES IN SHREDS = 100. Finally, placement and orientation errors exhibiting obvious discrete chromatin interaction patterns were manually adjusted. The total length of pseudochromosomes consisted of 93.25% of all genome sequences (Supplementary Figure 1 and Supplementary Tables 4, 5).
Genome annotation
The I. latifolia genome was annotated using genomic sequences as well as repeated sequences, gene structure information, non-coding RNAs, pseudogenes, and gene function information. For repeated sequences, a de novo repeat library was initially constructed using LTR FINDER v1.05 (Xu and Wang, 2007) and Repeat Scount v1.0.5 (Price et al., 2005). The repeat library was classified by PASTEClassifier (Hoede et al., 2014) and then merged with Repbase (Jurka et al., 2005). Repeated sequence annotation was conducted according to the homolog method by RepeatMasker (Chen, 2004) using the merged database (Supplementary Table 6). De novo repeat annotation was performed using RepeatModeller (Flynn et al., 2020). Three methods were selected to annotate gene structures. First, Genscan (Burge and Karlin, 1997), AUGUSTUS (Stanke et al., 2006), GlimmerHMM (Majoros et al., 2004), Gene ID (Alioto et al., 2018), and SNAP (Korf, 2004) were applied for de novo prediction according to the I. latifolia genome. Second, the protein sequences of three related species were selected for homologous annotation using GeMoMa (Keilwagen et al., 2019). Third, transcript annotations were performed based on the RNA sequencing results using HISAT (Kim et al., 2015), StringTie (Pertea et al., 2015), TransDecoder, GeneMarkS-T (Tang et al., 2015), and PASA (Campbell et al., 2006). Finally, all acquired data based on the three predictions were combined and revised using EVidenceModeler (EVM) (Haas et al., 2008) and PASA (Campbell et al., 2006; Supplementary Figure 2 and Supplementary Tables 7, 8). The annotations of the non-coding RNAs included microRNAs (miRNAs), ribosomal RNAs (rRNAs), and transfer RNAs (tRNAs). Infenal v1.1 (Nawrocki and Eddy, 2013) was used to annotate miRNAs and rRNAs based on the miRbase (Kozomara et al., 2019) and Rfam (Griffiths-Jones et al., 2005) databases, respectively. tRNAscan-SE (Lowe and Eddy, 1997) with the “-E -H” option was applied to detect the tRNA sequences. Pseudogene homolog sequences were subjected to BLAST searches using GenBlastA (She et al., 2009). The non-mature termination codes and frameshift mutations of pseudogenes were analyzed and annotated using GeneWise (Birney et al., 2004). Gene functions were annotated via protein databases, including the NCBInr, euKaryotic Orthologous Groups (KOGs) (Koonin et al., 2004), Gene Ontology (GO) (Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000), and SWISS-PROT/TrEMBL (Boeckmann et al., 2003) databases, using protein sequences whose structures had been annotated (Supplementary Table 9).
Comparative genomics and genome evolution analyses
Orthologous gene clusters of I. latifolia and 13 other angiosperms were identified using OrthoFinder (Emms and Kelly, 2019) with the default parameters. A total of 13,615 homologous groups were identified in I. latifolia, and 1,002 low-copy orthologous genes were identified in this set. The protein sequences of low-copy orthologous genes were aligned using MUSCLE v3.8.31 (Edgar, 2004) and were then used to build a highly supported maximum likelihood (ML) tree of 14 angiosperm species with the JTT + F + R5 best-fit model using IQ-TREE v1.6.11 (Nguyen et al., 2015). To further estimate the divergence times of I. latifolia and the other 13 angiosperm species, the MCMCTree program included in PAML v4.9i (Yang, 2007) was applied to calculate their divergence times. Four calibration points were selected from TimeTree3 as normal priors to reduce age, referencing speciation times of 168–194 Mya for the split of I. latifolia and Amborella trichopoda, 148–173 Mya for that of I. latifolia and Piper nigrum, 110–124 Mya for that of I. latifolia and Arabidopsis thaliana, and 88–106 Mya for that of I. latifolia and Panax notoginseng.
The analysis of the expansion and contraction of orthologous gene families in I. latifolia and 13 other angiosperm species was performed using the software CAFE 5 (Mendes et al., 2021). WGD software (Zwaenepoel and Van de Peer, 2019) was used for the analysis of synonymous substitutions per synonymous site (Ks) value-based paralog age distributions. All potential paralogs were detected via all-vs.-all protein sequence BLAST searches using BLASTP with an e-value cut-off of 10–10, and the MCL package was then used for gene family construction. MAFFT (Rozewicki et al., 2019) was used to align each family. Then, gene families (with n members) of n*(n-1)/2 > “max pairwise” were removed, and a phylogenetic tree was built for each gene family using FastTree (Price et al., 2009). Ks values were calculated using the maximum-likelihood method in the CODEML program of the PAML v.4.4c package (Yang, 2007). Finally, we performed mixture modeling for all possible WGD inferences using the BGMM method. The JCVI (Tang et al., 2008) and Minimap packages (Li, 2018) were used for syntenic visualization. The WGDI package (Sun et al., 2021) was used for collinear anchor pair identification and analysis. All syntenic blocks were identified using the improved collinearity pipeline in WGDI with the “p-value = 0.05” setting, and the Ks value for each anchor pair gene located in a syntenic block was calculated using the Ks pipeline in WGDI. The Ks dotplot of all anchor pairs was obtained by applying the block pipeline in WGDI. The KsPeaks pipeline in WGDI was used for the distribution analysis of the Ks median value for each syntenic block. Finally, all of the above results from the Ks distributions were summarized in one picture by plotting with the ggplot2 package.
Transcriptome sequencing and evolutionary analysis of gene families in Ilex
The leaves of I. latifolia and I. cornuta at five developmental stages and the fruits of I. latifolia at two developmental stages (green and red) were collected from plants cultivated on the campus of Nanjing Forestry University. Three replicate samples were selected for each stage. All samples were frozen in liquid nitrogen immediately after harvesting. The RNA of the samples was extracted using the RNA Plant Plus Kit (Tiangen, DP473) according to the manufacturer’s protocol. Illumina RNA-Seq libraries were prepared and sequenced on a HiSeq 2500 system following the manufacturer’s instructions (Illumina, United States). Raw reads were trimmed to remove adaptors, and short reads (<100 bp after trimming) were discarded. The TopHat2 package (Kim et al., 2013) was used to map clean reads to the genome with the default parameters. Transcripts were assembled using Cufflinks (Trapnell et al., 2012), and TransDecoder software4 was used for annotation. Gene expression levels were calculated and normalized using the FPKM method, followed by the application of RSEM software (Li and Dewey, 2011). Gene expression heatmaps were generated using TBtools (Chen et al., 2020).
HMMER (Finn et al., 2011) and BLASTP (Camacho et al., 2009) were used to identify putative P450 gene families from the protein sequences of I. latifolia and I. cornuta. The Hidden Markov Model (HMM) database with the seed file (PF00067) of the P450 genes and the P450 family proteins of Arabidopsis thaliana were obtained from the Arabidopsis Information Resource (TAIR).5 The filtered sequences were further subjected to BLAST searches using the NCBI database6 with a cut-off E-value of 10–5. Sequences annotated as P450 members in I. latifolia and I. cornuta were collected and aligned with those from Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, and Panax notoginseng using the ClustalW program (Oliver et al., 2005). An unrooted maximum-likelihood phylogenetic tree was then constructed using IQ-TREE (Nguyen et al., 2015) and visualized using Figtree7 and iTOL (Letunic and Bork, 2021). The identification of genes in the anthocyanin synthesis pathway following the same methods applied to the P450 gene family.
Results
Genome sequencing, assembly, and annotation
In this study, 71,218,217,963 k-mers were produced, and the peak depth of the k-mers was 89 (Supplementary Figure 3). The whole-genome size was approximately 772.55 Mb, which was close to the genome size estimated from flow cytometry (Su et al., 2020). The calculated repeat and heterozygosity rates were 47.59 and 0.85%, respectively (Supplementary Table 10). To obtain a high-quality genome, a combination of 88.6 G Illumina paired-end reads (120×), 82.4 G Nanopore single-molecule long reads (N50 = 33.1 Kb, 108×), and 83.8 G of Hi-C sequencing data (110×) were used for assembly. After genome assembly, polishing, and redundancy elimination, an initial assembly of 765.94 Mb with 844 contigs was obtained for the I. latifolia genome (contig N50 = 1.46 Mb), which was further assembled into 344 scaffolds (scaffold N50 = 33.4 Mb) (Supplementary Table 11). Altogether, 764.44 Mb (99.8%) of the assembly could be anchored to 20 pseudochromosomes (Figure 1B), and the genome-wide interaction heatmap showed high-quality grouping and ordering results based on the Hi-C data (Supplementary Figure 1).
Genome annotation indicated that the I. latifolia genome contained 406 Mb (53.0%) of repetitive sequences, among which LTR elements were the most abundant components (29.4%) (Supplementary Table 6). Regarding protein-coding genes, ab initio predictions incorporating the homology-based method and transcriptome data identified 35,218 genes in the I. latifolia genome, with an average coding-sequence length of 1.55 Kb and an average of 5.2 exons per gene. Among all annotated genes, 24,498 (69.6%) were supported by transcriptomic data, and 30,586 (86.8%) genes could be functionally annotated (Supplementary Figures 4, 5). Our annotation captured 1,328 (92.22%) complete benchmarking universal single-copy ortholog (BUSCO) genes, suggesting that our assembly achieved a high level of genome completeness. In addition to protein-coding genes, we identified 448 transfer RNAs, 314 ribosomal RNAs, and 127 microRNAs.
Phylogenetic position of Aquifoliales and whole-genome duplications in Ilex latifolia
Aquifoliales was stably recovered as the first diverging lineage of campanulids based on plastid data (Moore et al., 2010; Li et al., 2019); however, it repeatedly fell within lamiids in phylogenies reconstructed based on nuclear genes (Zeng et al., 2017; Yang et al., 2020). To resolve the phylogenetic position of Aquifoliales, we extracted 1,002 low-copy orthologous nuclear genes from 14 angiosperm species and reconstructed a highly supported phylogenetic tree (Figure 2A). Our results showed that the overall relationships of those 14 species were nearly identical to the backbone Angiosperm Phylogeny Group IV (APG IV, 2016), and I. latifolia was placed at the basal position of campanulids as the sister group to all other campanulids (BS = 100%), supporting the hypothesis that Aquifoliales belongs to campanulids. The divergence of Aquifoliales represent the initiation of campanulid differentiation, which was estimated to have started approximately 78 million years ago. Among all protein-coding genes, 6,644 gene families were shared by all 14 angiosperms, and 6,971 gene families were specific to I. latifolia (Figure 2B). In addition to the specific gene families, 2,991 gene families were found to have significantly expanded in I. latifolia, whereas 1,781 gene families significantly contracted (p < 0.05, Figure 2A). The specific and expanded gene families may have contributed to the development of the species-specific properties of this plant, leading to its distinct morphological, physiological, and genetic characteristics. GO analysis shows that the specific and expanded gene families in holly are associated with secondary metabolic processes, auxin catabolic process and other activities (Supplementary Figure 6), notably, KEGG analysis shows that both specific and expanded gene families related to terpene skeleton biosynthesis (Supplementary Figure 7).
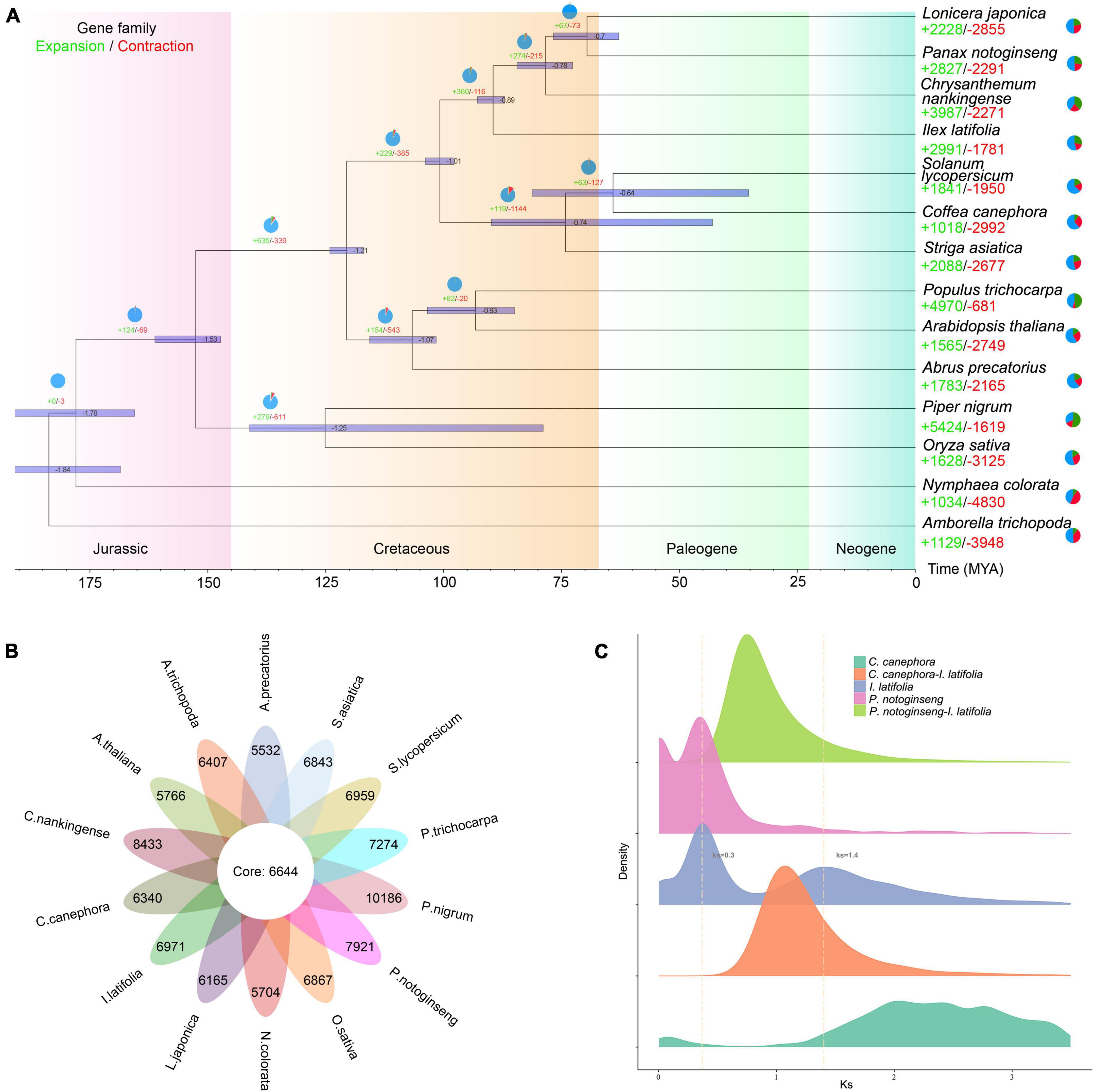
Figure 2. Genome evolution analysis of Ilex latifolia. (A) Expansion and contraction of gene families and phylogenetic relationships and divergence times between I. latifolia and other plant species. The light green numbers represent the numbers of expanded gene families, and the red numbers represent the numbers of contracted gene families. (B) The shared and unique gene families were compared between I. latifolia and 13 other angiosperm species. Each number represents the number of gene families. (C) Ks distribution and WGD events in I. latifolia. The Ks distribution of I. latifolia shows two peaks, one at approximately 0.3 (WGD 2) and another at approximately 1.4 (WGD 1).
Whole-genome duplication (WGD) events have commonly occurred among flowering plants (Van de Peer et al., 2017). To identify potential WGD events during the course of I. latifolia evolution, we calculated the Ks values of paralogs located in syntenic blocks of the I. latifolia genome. The resulting Ks distribution of I. latifolia showed two distinct peaks, one at approximately 0.3 (WGD 1) and the other at approximately 1.4 (WGD 2), suggesting that the ancestors of I. latifolia underwent at least two WGD events (Figure 2C and Supplementary Figures 8, 9). As studies of the other available genome from this genus, I. polyneura, have also identified two WGDs, with a similar Ks distribution pattern (Yao et al., 2022), we can speculate that the two WGDs likely occurred in the common ancestor of the two Ilex species. The more ancient WGD (WGD2) showed a peak before the divergence of I. latifolia and coffee, which would therefore correspond to the polyploidy event shared by all eudicots. The synteny analysis between I. latifolia and grape verified this conclusion by showing a 2:1 ratio of syntenic blocks (Supplementary Figure 9), which is expected to be a result of WGD1 in I. latifolia, and I. latifolia and grape should also share WGD2. The observation that the WGD2 peak of I. latifolia did not coincide with the peak in coffee can likely be explained by the differential evolutionary rates of the two species. For WGD1, although Panax notoginseng also showed a peak at 0.3, this Araliaceae species likely experienced an independent WGD, different from that in I. latifolia, because P. notoginseng and I. latifolia diverged earlier than the two independent WGDs. These findings suggest the occurrence of frequent and independent polyploidization events among different angiosperm lineages and the intensive occurrence of such events at certain geological times during angiosperm evolution.
Biosynthesis of pentacyclic triterpenoid saponins
Triterpenes constitute a large and structurally diverse class of natural products with considerable industrial and pharmaceutical value. The biosynthetic process involves a series of enzymes. To date, the biosynthesis of precursors and the corresponding enzymes have been identified (Sawai and Saito, 2011; Miettinen et al., 2017). However, the final steps are much more complicated among different taxa. Three types of enzymes–Oxidosqualene cyclases (OSC), cytochrome P450 monooxygenases (CYP450) and uridine diphosphate-dependent glycosyltransferases (UGT) are considered to be involved in the final steps, among which, CYP450-catalyzed structural modifications are the most critical and determine the diversification and functionalization of the triterpene scaffolds (Ghosh, 2017). CYP450 is the largest family of enzymes involved in plant metabolism, and some of its members can catalyze the formation of derivatives from basic triterpene skeletons with modified structures and various functions. The members of the CYP51H, CYP71A, D, CYP72A, CYP81Q, CYP87D, CYP88D, L, CYP89A, CYP93E, CYP705A, CYP708A, and CYP716A, C, E, S, U, and Y subfamilies are reported to be responsible for the biosynthesis and structural modification of triterpenes and related derivatives (Miettinen et al., 2017; Zheng et al., 2019).
To identify CYP450 genes involved in the biosynthesis of pentacyclic triterpenoid saponins in I. latifolia, we performed a genome-wide search for CYP450 genes in I. latifolia, Amborella trichopoda, Arabidopsis thaliana, Oryza sativa, and P. notoginseng by using BLAST and HMMER and conducted a phylogenetic analysis using the identified genes. The phylogenetic analysis indicated that all CYP450 genes could be classified into four monophyletic groups (i, ii, iii, and iv). The I. latifolia genome encodes 417 CYP450 genes, including four subfamilies with possible relationships to triterpene biosynthesis—CYP71A, CYP72A, CYP89A, and CYP716A—distributed in Groups i, ii, and iii (Figure 3A). Among the four subfamilies, three appear to have expanded compared with their numbers in P. notoginseng (another campanulid species): CYP71A (10 members), CYP72A (10 members), and CYP716A (22 members) (Supplementary Table 12). We further sequenced the transcriptomes of I. latifolia leaves at five developmental stages (three replicates for each stage) and examined the expression of these CYP450 genes using transcriptomic data. We found that two CYP716A genes (Ila16G023040.1 and Ila16G023050.1) and one CYP72A gene (Ila16G010270.1) were always highly expressed at all five stages, suggesting that they probably play important roles in the biosynthetic pathway of the I. latifolia pentacyclic triterpenoid saponins (Figure 3B).
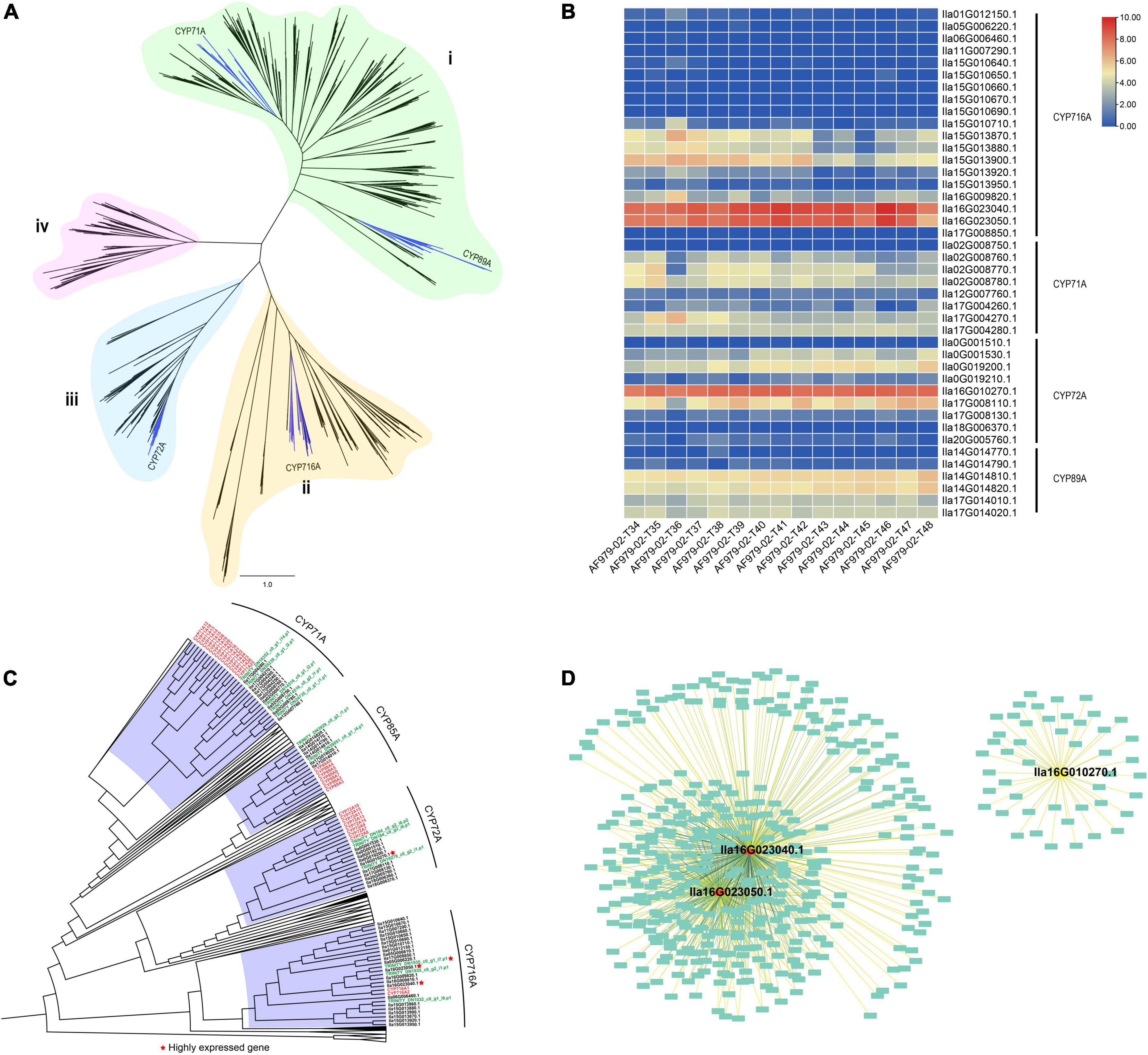
Figure 3. Transcriptomes of Ilex latifolia leaves at five developmental stages reveal the biosynthesis of pentacyclic triterpenoid saponins. (A) Phylogeny of 395 putative functional CYP450 genes from I. latifolia. (B) Heatmap of CYP450 gene expression related to triterpenoid synthesis. (C) A simplified CYP450 phylogenetic tree for Amborella trichopoda, Arabidopsis thaliana, I. latifolia, I. cornuta, and Oryza sativa. (D) Sub-network for three highly expressed CYP450 genes lla16G023040.1, lla16G023050.1, and lla16G010270.1.
Ilex latifolia is preferentially used as the material for making Kudingcha over other species because of the higher levels of pentacyclic triterpenoid saponins levels in its leaves, and our metabolomics study also identified higher pentacyclic triterpenoid saponin concentrations in I. latifolia than in its close relative I. cornuta (Supplementary Figure 10). We infer that this difference should be attributed to both different CYP450 gene numbers and their differential expression in the two species (Supplementary Figure 11). There were significantly fewer CYP71A (5 members), CYP72A (3 members) and CYP716A (3 members) genes in I. cornuta (Supplementary Table 12) than in I. latifolia, and detailed observations revealed that no orthologs of the highly expressed I. latifolia CYP72A (Ila16G010270.1) and CYP716A (Ila16G023040.1) genes were detected in any of the I. cornuta transcriptomes (Figure 3C), suggesting the absence of these critical genes in I. cornuta. Therefore, it is likely that the absence of critical CPY450 genes in I. cornuta is responsible for the low pentacyclic triterpenoid saponin concentration in this species and explains why the leaves of I. latifolia are favored for the production of Kudingcha over other species in the same genus.
Coexpression analysis using the WGCNA approach was then conducted to identify more potential genes involved in the process. According to the observed expression patterns, I. latifolia genes were classified into 44 modules (Supplementary Figure 12). The highly expressed CYP716A and CYP72A genes were classified into two modules (black and light green). The two modules comprised 886 (black) and 369 (light green) genes showing similar expression patterns to CYP716A and CYP72A genes, respectively (Figure 3D). The GO analysis indicated that 41 genes in the black module were related to oxidoreductase activity, acting on paired donors with the incorporation or reduction of molecular oxygen (Supplementary Figure 13), and there were 47 genes in the light green module associated with metal ion binding (Supplementary Figure 14). Therefore, these genes are also likely to be involved in the biosynthesis of pentacyclic triterpenoid saponins.
The biosynthesis of anthocyanidins regulates the red fruit color of I. latifolia and determines its ornamental value
Red fruits are the most appreciated ornamental characteristic of I. latifolia. The red color of fruit pericarp is usually regulated by the synthesis of anthocyanidins, specifically the elevated synthesis of pelargonidin and cyanidin. To examine whether the green-to-red color change in I. latifolia fruits was related to the increased production of pelargonidin and cyanidin, we identified genes in the biosynthesis pathway of pelargonidin and cyanidin and examined their expression in green and red fruit pericarps, respectively. Most genes showed higher expression in red fruit pericarps than in green pericarps (Figure 4A and Supplementary Figure 15). To more precisely determine the expressional differences in these genes in the green and red fruit pericarps, we conducted an qRT–PCR analysis, and the results showed an average 1.07-fold increase in the expression of genes in the biosynthesis pathway of pelargonidin and cyanidin (Supplementary Figure 16), among which F3H (Ila10G001350.1) and F3’H (Ila14G014210.1) showed the largest increases in the red pericarp (>1.4-fold) (Figure 4B). These results indicate that the synthesis of pelargonidin and cyanidin plays an important role in the I. latifolia fruit pericarp color change.
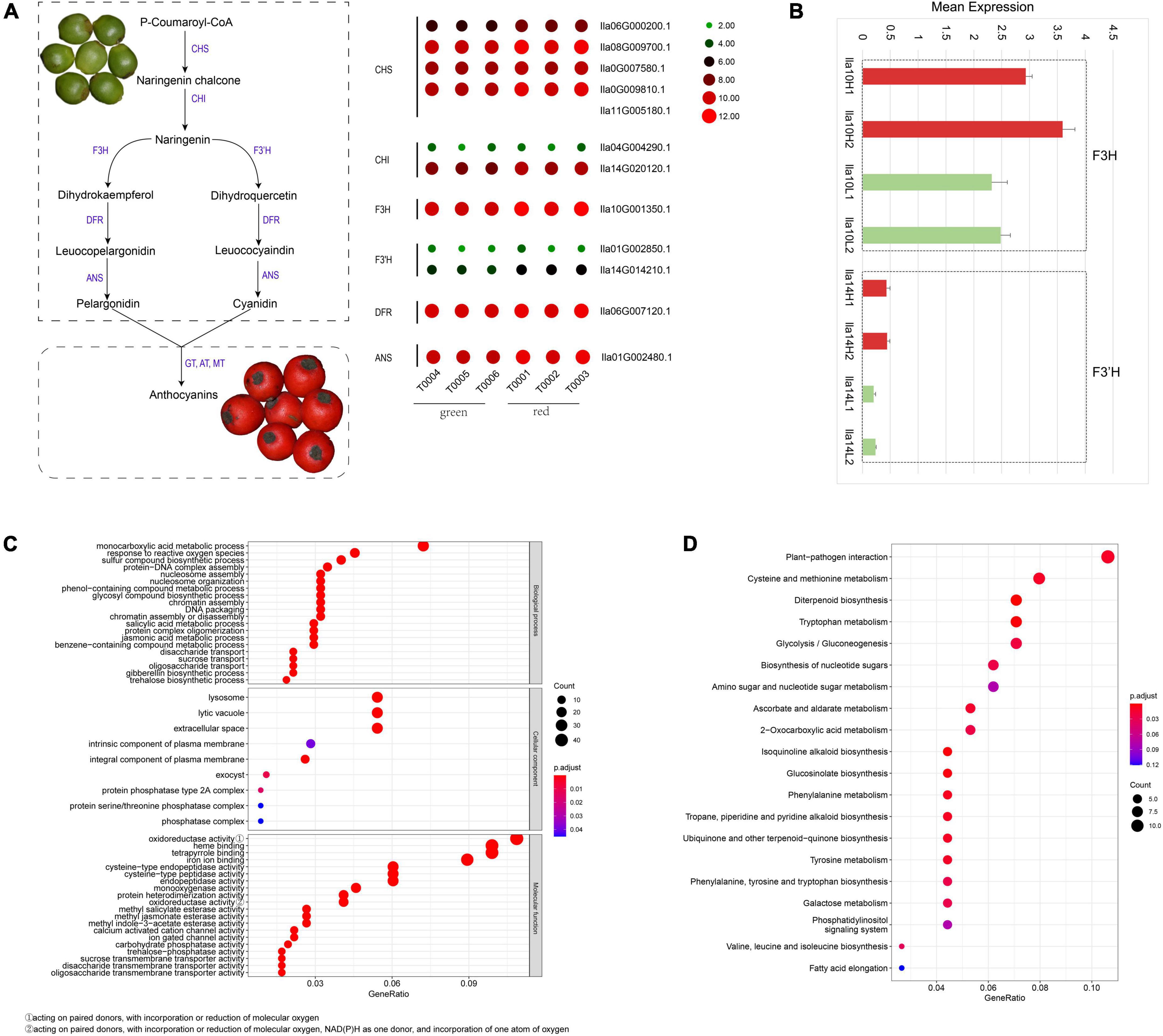
Figure 4. Anthocyanidin biosynthesis pathways in the Ilex latifolia fruit pericarp. (A) Genes in the biosynthesis pathway of pelargonidin and cyanidin and their expression in green and red fruit pericarps, respectively. (B) The expression of F3H (Ila10G001350.1) and F3’H (Ila14G014210.1) genes in the biosynthesis pathway of pelargonidin and cyanidin in the red (indicated by red histograms) and green (indicated by green histograms) pericarp using qRT–PCR analysis. (C) GO functional enrichment analysis of genes with significantly differential expression in I. latifolia. (D) KEGG analysis of genes with significantly differential expression in I. latifolia.
In addition to the genes in the biosynthesis pathway of pelargonidin and cyanidin, other genes may also be related to the fruit pericarp color change in I. latifolia. We identified 161 genes with significantly differential expression in the transcriptomes of green and red fruit pericarps. GO analysis indicated that genes related to the “monocarboxylic acid metabolic process” term were the most enriched in the “biological process” category; “lysosome,” “lytic vacuole,” and “extracellular space” were most enriched terms in the “cellular component” category; and “oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen,” “heme binding,” “tetrapyrrole binding,” and “tetrapyrrole binding” were the most significant terms in the “molecular function” category (Figure 4C). KEGG analysis indicated that the maturation of the fruit pericarp is most tightly connected with the improvement of pathogen immunity, followed by metabolism, including cysteine and methionine metabolism, diterpenoid biosynthesis, tryptophan metabolism, and glycolysis/gluconeogenesis, suggesting a complex process of fruit pericarp development (Figure 4D).
Discussion
The rapid development of genome sequencing technology has created the opportunity to acquire higher-quality genomes of important woody plant species with important ornamental and economic value. However, most of the angiosperm genomes sequenced to date have come from herbaceous plants (Kersey, 2019). Reference genomes for most woody plant families, which usually include important ornamental and economic tree species, are still lacking. Hollies, comprising more than 600 species worldwide, constitute one of the largest woody genera in Aquifoliaceae (Loizeau et al., 2016). However, research on these plants is currently hampered by the lack of reference genomes. In this study, we provide a chromosome-level assembly for I. latifolia, a species that can be used for beverage production, medicinal and ornamental purposes, and we used these data combined with multiomics data to explore the molecular basis underlying these features.
Ilex latifolia has multiple uses: its young leaves are employed for making healthy tea known as Kudingcha, and its evergreen leaves and red fruits are employed for ornamental purposes. The high-quality assembled genome of I. latifolia could provide much information for exploring questions related to the evolution of this species, the biosynthesis of the key components of Kudingcha, and the mechanism underlying its ornamental characteristics. Systematically, I. latifolia belongs to Aquifoliales, and this order mainly comprises woody trees and shrubs with sawtooth leaves and drupes. Our phylogenetic analysis recovered I. latifolia as the first diverging lineage of campanulids, consistent with the inferences made based on plastid genes (Moore et al., 2010; Li et al., 2019) but incongruent with the results based on nuclear genes (Zeng et al., 2017; Yang et al., 2020). Nevertheless, the branches splitting campanulids and lamiids are very short, suggesting that the campanulid-lamiid divergence was likely a rapid process and that Aquifoliales originated early, shortly after this divergence, as reflected in its own short branch length. Therefore, the plastid-nuclear conflict in the positioning of Aquifoliales should imply ancient hybridizations upon the early evolution of asterids, and the incongruence between our result and other studies based on nuclear genes may suggest potential incomplete lineage sorting. Hence, further evidence, such as genomic structures or phylogenies based on mitochondrial genes, is still required to conclusively determine the phylogenetic position of Aquifoliales.
The health care and medicinal effect of Kudingcha can likely be mainly attributed to the enrichment of pentacyclic triterpenoid saponins in the leaves of I. latifolia. Although the synthesis pathway of triterpenes has been well studied, the enzymes responsible for further modification leading to the generation of pentacyclic triterpenoid saponins remain poorly understood (Miettinen et al., 2017; Zheng et al., 2019). In this study, we screened potential enzymes based on a combination of genome-wide identification, phylogenomic analysis, and comparative transcriptomic approaches. Three members of the CYP450 superfamily belonging to the 72A and 716A subfamilies were suggested to be the most promising candidates according to phylogenomic clues and expressional patterns. Using the three candidate genes as hub genes, we performed WGCNA to identify more potential genes involved in the biosynthesis of pentacyclic triterpenoid saponins through coexpression networks. Our analytical strategy successfully identified two critical genes and a series of genes potentially involved in the biosynthesis of pentacyclic triterpenoid saponins. These identified genes will provide valuable information and will serve as targets for functional characterization in the future. Our study will greatly accelerate the elucidation of the whole biosynthesis pathway of this important metabolic product in I. latifolia and generate more interest in Kudingcha.
Anthocyanins found in plant organs commonly play an important role as an indicator of the ornamental value of plants (Li et al., 2020). As a fruit plant included in gardens, I. latifolia bears multiple red decorative berries in its ripening season and contributes to a very beautiful landscape. However, the mechanism of color change in I. latifolia fruit pericarps is still unknown. Therefore, the genes in the biosynthesis pathway of pelargonidin and cyanidin were identified in this study, and their expression in green and red fruit pericarps was examined. Interestingly, most of these genes were more highly expressed in red fruit pericarps than in green pericarps. This finding revealed that the synthesis of pelargonidin and cyanidin also plays an important role in the process of I. latifolia fruit pericarp color change. In addition, there were 161 genes with significantly differential expression in the transcriptomes of the two developmental stages of the fruit pericarp. Both GO and KEGG analyses indicate that fruit pericarp development is a complex process, and more studies are still needed to reveal the mechanism of fruit pericarp development.
In summary, the high-quality I. latifolia reference genome combined with transcriptome data provided insights into genome evolution, the biosynthesis of the pentacyclic saponin triterpenes found in Kudingcha, and the biosynthesis of anthocyanidins in the fruit pericarp of I. latifolia. The genome and transcriptome data obtained in this study will also be useful for studies concerning the production of holly teas and medicines, the mechanisms underlying the formation of important ornamental traits and molecular breeding in I. latifolia and other Ilex species.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://ngdc.cncb.ac.cn/gwh, GWHBIST00000000.
Author contributions
K-WX, Y-FD, and J-YX conceived and designed the project. K-WX, C-XL, QZ, MZ, PZ, Y-MF, and Y-FD collected and generated the plant materials. K-WX, MZ, and X-FW performed all the data analyses under the supervision of J-YX, Y-FD, and Y-MF. K-WX, X-FW, and J-YX drafted the manuscript. All authors contributed to and approved the final manuscript.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province (#BK20210612), the National Natural Science Foundation of China (#32100167), the Nanjing Agricultural University project funding (#KYCXJC2022003), and the Nanjing Forestry University project funding (#163108093).
Acknowledgments
The computational resources and services were provided by the Bioinformatics Center of Nanjing Agricultural University. We thank the editor and the other reviewers for their helpful comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.982323/full#supplementary-material
Footnotes
- ^ https://github.com/ruanjue/smartdenovo
- ^ https://github.com/nservant/HiC-Pro
- ^ http://www.timetree.org
- ^ https://github.com/TransDecoder/TransDecoder/releases
- ^ http://www.Arabidopsis.org/
- ^ https://www.ncbi.nlm.nih.gov/
- ^ http://tree.bio.ed.ac.uk/software/figtree
References
Alioto, T., Blanco, E., Parra, G., and Guigó, R. (2018). Using geneid to identify genes. Curr. Bioinform. 64:e56. doi: 10.1002/cpbi.56
APG IV (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bracesco, N., Sanchez, A. G., Contreras, V., Menini, T., and Gugliucci, A. (2011). Recent advances on Ilex paraguariensis research: Minireview. J. Ethnopharmacol. 136, 378–384. doi: 10.1016/j.jep.2010.06.032
Belton, J. M., McCord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., and Dekker, J. (2012). Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276. doi: 10.1016/j.ymeth.2012.05.001
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M., and Buell, C. R. (2006). Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7:327. doi: 10.1186/1471-2164-7-327
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chen, N. (2004). Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4–10. doi: 10.1002/0471250953.bi0410s05
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. P. Natl. Acad. Sci. U.S.A. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Folch, C. (2021). Ceremony, medicine, caffeinated Tea: Unearthing the forgotten faces of the North American stimulant yaupon (Ilex vomitoria). Comp. Stud. Soc. Hist. 63, 464–498. doi: 10.1017/S0010417521000116
Ghosh, S. (2017). Triterpene structural diversification by plant cytochrome P450 enzymes. Front. Plant Sci. 8:1886. doi: 10.3389/fpls.2017.01886
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124. doi: 10.1093/nar/gki081
Gottlieb, A. M., Giberti, G. C., and Poggio, L. (2005). Molecular analyses of the genus Ilex (Aquifoliaceae) in southern South America, evidence from AFLP and ITS sequence data. Am. J. Bot. 92, 352–369. doi: 10.3732/ajb.92.2.352
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Boil. 9, 1–22. doi: 10.1186/gb-2008-9-1-r7
Hoede, C., Arnoux, S., Moisset, M., Chaumier, T., Inizan, O., Jamilloux, V., et al. (2014). PASTEC: An automatic transposable element classification tool. PLoS One 9:e91929. doi: 10.1371/journal.pone.0091929
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Koonin, E. V., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Krylov, D. M., Makarova, K. S., et al. (2004). A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Boil. 5, 1–28.
Keilwagen, J., Hartung, F., and Grau, J. (2019). “GeMoMa: Homology-based gene prediction utilizing intron position conservation and RNA-seq data,” in Gene Prediction, ed. M. Kollmar (New York, NY: Humana), 161–177. doi: 10.1007/978-1-4939-9173-0_9
Kersey, P. J. (2019). Plant genome sequences: Past, present, future. Curr. Opin. Plant Biol. 48, 1–8. doi: 10.1016/j.pbi.2018.11.001
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Boil. 14, 1–13. doi: 10.1186/gb-2013-14-4-r36
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. methods 12, 357–360. doi: 10.1038/nmeth.3317
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: From microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. doi: 10.1093/nar/gky1141
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, B., and Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, L., Xu, L. J., Ma, G. Z., Dong, Y. M., Peng, Y., and Xiao, P. G. (2013). The large-leaved Kudingcha (Ilex latifolia Thunb and Ilex kudingcha CJ Tseng): A traditional Chinese tea with plentiful secondary metabolites and potential biological activities. J. Nat. Med. 67, 425–437. doi: 10.1007/s11418-013-0758-z
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Yang, Z., Zeng, Q., Wang, S., Luo, Y., Huang, Y., et al. (2020). Abnormal expression of bHLH3 disrupts a flavonoid homeostasis network, causing differences in pigment composition among mulberry fruits. Hortic Res. 7:83. doi: 10.1038/s41438-020-0302-8
Li, H. (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H. T., Yi, T. S., Gao, L. M., Ma, P. F., Zhang, T., Yang, J. B., et al. (2019). Origin of angiosperms and the puzzle of the jurassic gap. Nat. Plants 5, 461–470. doi: 10.1038/s41477-019-0421-0
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Loizeau, P. A., Savolainen, V., Andrews, S., Barriera, G., and Spichiger, R. (2016). “Aquifoliaceae,” in Flowering plants. Eudicots. The families and genera of vascular plants, eds J. W. Kadereit and C. Jeffrey (Berlin: Springer), 31–36. doi: 10.1007/978-3-319-28534-4_3
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Manen, J. F., Barriera, G., Loizeau, P. A., and Naciri, Y. (2010). The history of extant Ilex species (Aquifoliaceae): Evidence of hybridization within a Miocene radiation. Mol. Phylogenet. Evol. 57, 961–977. doi: 10.1016/j.ympev.2010.09.006
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Mendes, F. K., Vanderpool, D., Fulton, B., and Hahn, M. W. (2021). CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518. doi: 10.1093/bioinformatics/btaa1022
Miettinen, K., Pollier, J., Buyst, D., Arendt, P., Csuk, R., Sommerwerk, S., et al. (2017). The ancient CYP716 family is a major contributor to the diversification of eudicot triterpenoid biosynthesis. Nat. Commun. 8, 1–13. doi: 10.1038/ncomms14153
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. P. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Bio. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Oliver, T., Schmidt, B., Nathan, D., Clemens, R., and Maskell, D. (2005). Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics 21, 3431–3432. doi: 10.1093/bioinformatics/bti508
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Bio. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Rozewicki, J., Li, S., Amada, K. M., Standley, D. M., and Katoh, K. (2019). MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 47, W5–W10. doi: 10.1093/nar/gkz342
Sawai, S., and Saito, K. (2011). Triterpenoid biosynthesis and engineering in plants. Front. Plant Sci. 2:25. doi: 10.3389/fpls.2011.00025
Selbach-Schnadelbach, A., Cavalli, S. S., Manen, J. F., Coelho, G. C., and De Souza-Chies, T. T. (2009). New information for Ilex phylogenetics based on the plastid psbA-trnH intergenic spacer (Aquifoliaceae). Bot. J. Linn. Soc. 159, 182–193. doi: 10.1111/j.1095-8339.2008.00898.x
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C. J., Vert, J. P., et al. (2015). HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11. doi: 10.1186/s13059-015-0831-x
She, R., Chu, J. S. C., Wang, K., Pei, J., and Chen, N. (2009). GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 19:143. doi: 10.1101/gr.082081.108
Shi, L., Li, N. W., Wang, S. Q., Zhou, Y. B., Huang, W. J., Yang, Y. C., et al. (2016). Molecular evidence for the hybrid origin of Ilex dabieshanensis (Aquifoliaceae). PLoS One 11:e0147825. doi: 10.1371/journal.pone.0147825
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Su, T., Zhang, M. R., Shan, Z. Y., Li, X. D., Zhou, B. Y., Wu, H., et al. (2020). Comparative survey of morphological variations and plastid genome sequencing reveals phylogenetic divergence between four endemic Ilex species. Forests 11:964. doi: 10.3390/f11090964
Sun, P., Jiao, B., Yang, Y., Shan, L., Li, T., Li, X., et al. (2021). WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. BioRxiv [Preprint] doi: 10.1101/2021.04.29.441969
Sun, Y., Xu, W. Q., Zhang, W. Q., Hu, Q. H., and Zeng, X. X. (2011). Optimizing the extraction of phenolic antioxidants from kudingcha made from Ilex kudingcha C. J. Tseng by using response surface methodology. Sep. Purif. Technol. 78, 311–320. doi: 10.1016/j.seppur.2011.01.038
Tang, S., Lomsadze, A., and Borodovsky, M. (2015). Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43, e78–e78. doi: 10.1093/nar/gkv227
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Van de Peer, Y., Mizrachi, E., and Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. doi: 10.1038/nrg.2017.26
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Vurture, G. W., Sedlazeck, F. J., Nattestad, M., Underwood, C. J., Fang, H., Gurtowski, J., et al. (2017). GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204. doi: 10.1093/bioinformatics/btx153
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9:e112963. doi: 10.1371/journal.pone.0112963
Xu, Z., and Wang, H. (2007). LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Xu, K. W., Lin, C. X., Lee, S. Y., Mao, L. F., and Meng, K. K. (2021). Comparative chloroplast genome analyses of Ilex (Aquifoliaceae): Insights into evolutionary dynamics and phylogenetic relationships. BMC Genomics 23:203. doi: 10.1186/s12864-022-08397-9
Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, L., Su, D., Chang, X., Foster, C. S., Sun, L., Huang, C., et al. (2020). Phylogenomic insights into deep phylogeny of angiosperms based on broad nuclear gene sampling. Plant Commun. 1:100027. doi: 10.1016/j.xplc.2020.100027
Yang, B., Yang, T., Tan, Q. L., Zhu, J. P., Zhou, L., Xiong, T. Q., et al. (2015). Antiplatelet aggregation triterpene saponins from the leaves of Ilex kudingcha. Phytochem. Lett. 13, 302–307. doi: 10.1016/j.phytol.2015.07.008
Yao, X., Song, Y., Yang, J. B., Tan, Y. H., and Corlett, R. T. (2021). Phylogeny and biogeography of the hollies (Ilex L. Aquifoliaceae). J. Syst. Evol. 59, 73–82. doi: 10.1111/jse.12567
Yao, X., Lu, Z. Q., Song, Y., Hu, X. D., and Corlett, R. T. (2022). A chromosome-scale genome assembly for the holly (Ilex polyneura) provides insights into genomic adaptations to elevation in Southwest China. Hortic. Res. 9:uhab049. doi: 10.1093/hr/uhab049
Zeng, L., Zhang, N., Zhang, Q., Endress, P. K., Huang, J., and Ma, H. (2017). Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 214, 1338–1354. doi: 10.1111/nph.14503
Zheng, X. Y., Li, P., and Lu, X. (2019). Research advances in cytochrome P450-catalysed pharmaceutical terpenoid biosynthesis in plants. J. Exp. Bot. 70, 4619–4630. doi: 10.1093/jxb/erz203
Keywords: Aquifoliales, whole-genome sequencing, genome evolution, pentacyclic triterpenoid saponins, anthocyanidins biosynthesis genes, holly
Citation: Xu K-W, Wei X-F, Lin C-X, Zhang M, Zhang Q, Zhou P, Fang Y-M, Xue J-Y and Duan Y-F (2022) The chromosome-level holly (Ilex latifolia) genome reveals key enzymes in triterpenoid saponin biosynthesis and fruit color change. Front. Plant Sci. 13:982323. doi: 10.3389/fpls.2022.982323
Received: 30 June 2022; Accepted: 04 August 2022;
Published: 22 August 2022.
Edited by:
Liangsheng Zhang, Zhejiang University, ChinaReviewed by:
Muhammad Aamir Manzoor, Anhui Agricultural University, ChinaYifei Liu, Hubei University of Chinese Medicine, China
Copyright © 2022 Xu, Wei, Lin, Zhang, Zhang, Zhou, Fang, Xue and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-Yu Xue, xuejy@njau.edu.cn; Yi-Fan Duan, yifanduan@njfu.edu.cn
†These authors have contributed equally to this work