 Yu Feng
Yu Feng Xin-Fen Gao
Xin-Fen Gao Jun-Yi Zhang
Jun-Yi Zhang Li-Sha Jiang
Li-Sha Jiang Xiong Li
Xiong Li Heng-Ning Deng
Heng-Ning Deng Min Liao
Min Liao Bo Xu
Bo Xu- 1CAS Key Laboratory of Mountain Ecological Restoration and Bioresource Utilization and Ecological Restoration and Biodiversity Conservation Key Laboratory of Sichuan Province, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, China
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 3Mangkang Ecological Station, Tibet Ecological Safety Monitor Network, Changdu, China
The genus Campylotropis Bunge (Desmodieae, Papilionoideae) comprises about 37 species distributed in temperate and tropical Asia. Despite the great potential in soil conservation, horticulture, and medicine usage, little is known about the evolutionary history and phylogenetic relationships of Campylotropis due to insufficient genetic resources. Here, we sequenced and assembled 21 complete chloroplast genomes of Campylotropis species. In combination with the previously published chloroplast genomes of C. macrocarpa and closely related species, we conducted comparative genomics and phylogenomic analysis on these data. Comparative analysis of the genome size, structure, expansion and contraction of inverted repeat (IR) boundaries, number of genes, GC content, and pattern of simple sequence repeats (SSRs) revealed high similarities among the Campylotropis chloroplast genomes. The activities of long sequence repeats contributed to the variation in genome size and gene content in Campylotropis chloroplast genomes. The Campylotropis chloroplast genomes showed moderate sequence variation, and 13 highly variable regions were identified for species identification and further phylogenetic studies. We also reported one more case of matK pseudogene in the legume family. The phylogenetic analysis confirmed the monophyly of Campylotropis and the sister relationship between Lespedeza and Kummerowia, the latter two genera were then sister to Campylotropis. The intrageneric relationships of Campylotropis based on genomic scale data were firstly reported in this study. The two positively selected genes (atpF and rps19) and eight fast-evolving genes identified in this study may help us to understand the adaptation of Campylotropis species. Overall, this study enhances our understanding of the chloroplast genome evolution and phylogenetic relationships of Campylotropis.
Introduction
The genus Campylotropis Bunge belongs to the tribe Desmodieae (Benth.) Hutchinson in the legume subfamily Papilionoideae. It comprises c. 37 species of deciduous shrubs and subshrubs that distributed in Asia from the Himalaya region through Southeast Asia to China and Korea (Barham, 1997; Iokawa and Ohashi, 2008; Huang et al., 2010). Southwest China is the diversity center of Campylotropis as it harbors c. 80% of the species, and c. 20 species are endemic to this region (Iokawa and Ohashi, 2008). Most species in this genus have important value in soil conservation due to their tolerance of arid soils (Huang et al., 2010). Some Campylotropis species are also valuable for horticulture and medicine usage. For example, C. polyantha is widely used in gardening due to its numerous racemes of showy flowers and long-lasting fluorescence (Barham, 1997). The dried roots of C. hirtella can be used as traditional Chinese medicine for the treatment of benign prostate hyperplasia (Wen et al., 2007), and C. trigonoclada contains daucosterol linoleate which can be used for the treatment of breast cancer (Han et al., 2018).
As suggested by previous molecular phylogenetic studies, Campylotropis is sister to the other two genera of subtribe Lespedezinae (i.e., Lespedeza and Kummerowia) in tribe Desmodieae (Xu et al., 2012; Jabbour et al., 2018; Jin et al., 2019). Much effort has been made to clarify species relationships within Campylotropis, mostly based on morphological characters such as leaf and calyx morphology (e.g., Iokawa and Ohashi, 2008; Huang et al., 2010). However, most of the morphological characteristics (e.g., persistence of bracts, the color of flowers, and shape of keel petals) are polymorphic and vary continuously among species, causing controversial species delimitation in this genus (Iokawa and Ohashi, 2008). Besides, little is known about its intrageneric and interspecific relationships due to the lack of comprehensive molecular phylogenetic studies.
Chloroplasts, derived from photosynthetic bacteria, play critical roles in the survival, adaptation, and evolution of plants (Wicke et al., 2011; Zhao et al., 2019; Dopp et al., 2021). Although the chloroplast (cp) genomes are much smaller than most nuclear genomes, they encode essential proteins related to photosynthesis, fixation of carbon and nitrogen, and biosynthesis of starch, pigments, fatty acids, and amino acids (Howe et al., 2003; Wicke et al., 2011; Daniell et al., 2016). Chloroplast genomes have relatively stable structure and gene content compared to nuclear genomes. The typical structure of angiosperm cp genome is a circular double-stranded DNA molecule, exhibiting a conserved quadripartite structure [i.e., two inverted repeats (IRs) separated by a large single-copy region (LSC) and a small single-copy region (SSC)] and containing 110–130 genes (Sugiura, 1992; Daniell et al., 2016). The characteristics of cp genomes including lack of recombination, low nucleotide substitution rates, and usually uniparental inheritance make them the primary source to explore phylogenetic evolution of plant species (Shaw et al., 2005). Besides, structural variants such as expansion and contraction of IRs, gains or losses of genes and introns, and dynamics of repeat sequences (e.g., simple sequence repeat, SSR) provide resources for evaluating genomic evolutionary history (e.g. Sabir et al., 2014; Keller et al., 2017). The development of sequencing technology and analysis tools makes the acquisition of cp genomes much easier than before, thus promptly extending gene-based phylogenetics to phylogenomics (Lu et al., 2017). In fact, recent phylogenomic studies have been successful in reconstructing phylogenies at various taxonomic scales (e.g., genera and families) across angiosperms using the cp genome datasets (e.g., Cai et al., 2015; Ruhsam et al., 2015; Luo et al., 2016; Zhang et al., 2017, 2021).
Here, we present 21 complete cp genomes of Campylotropis species assembled from Illumina short reads. In combination with the previously published cp genomes of C. macrocarpa (Jin et al., 2019) and closely related species, we conducted comparative genomics and phylogenomic analyses on these data with the following aims: (1) to reveal the global structural patterns of Campylotropis cp genomes; (2) to investigate variations of SSRs and repeat sequences among Campylotropis cp genomes; (3) to screen highly variable regions suitable for species identification and phylogenetic studies; (4) to reconstruct a robust phylogenetic relationship within Campylotropis and among genera in the tribe Desmodieae; and (5) to investigate adaptive evolution patterns of cp genes in Campylotropis. These results will provide insights into the evolutionary history of Campylotropis and tribe Desmodieae as well as abundant information for future phylogenetic and population genetic studies.
Materials and Methods
Taxon Sampling, DNA Extraction, and Sequencing
In this study, leaf materials of 21 accessions representing 17 Campylotropis species (including four subspecies, one variety, and one forma) were collected from the field and preserved in silica gel (Table 1). Voucher specimens were deposited in the Herbarium of the Chengdu Institute of Biology (CDBI; Supplementary Table S1). The extraction of total genomic DNA, library preparation, and Illumina sequencing for each accession were described in our previous study (Liao et al., 2021).
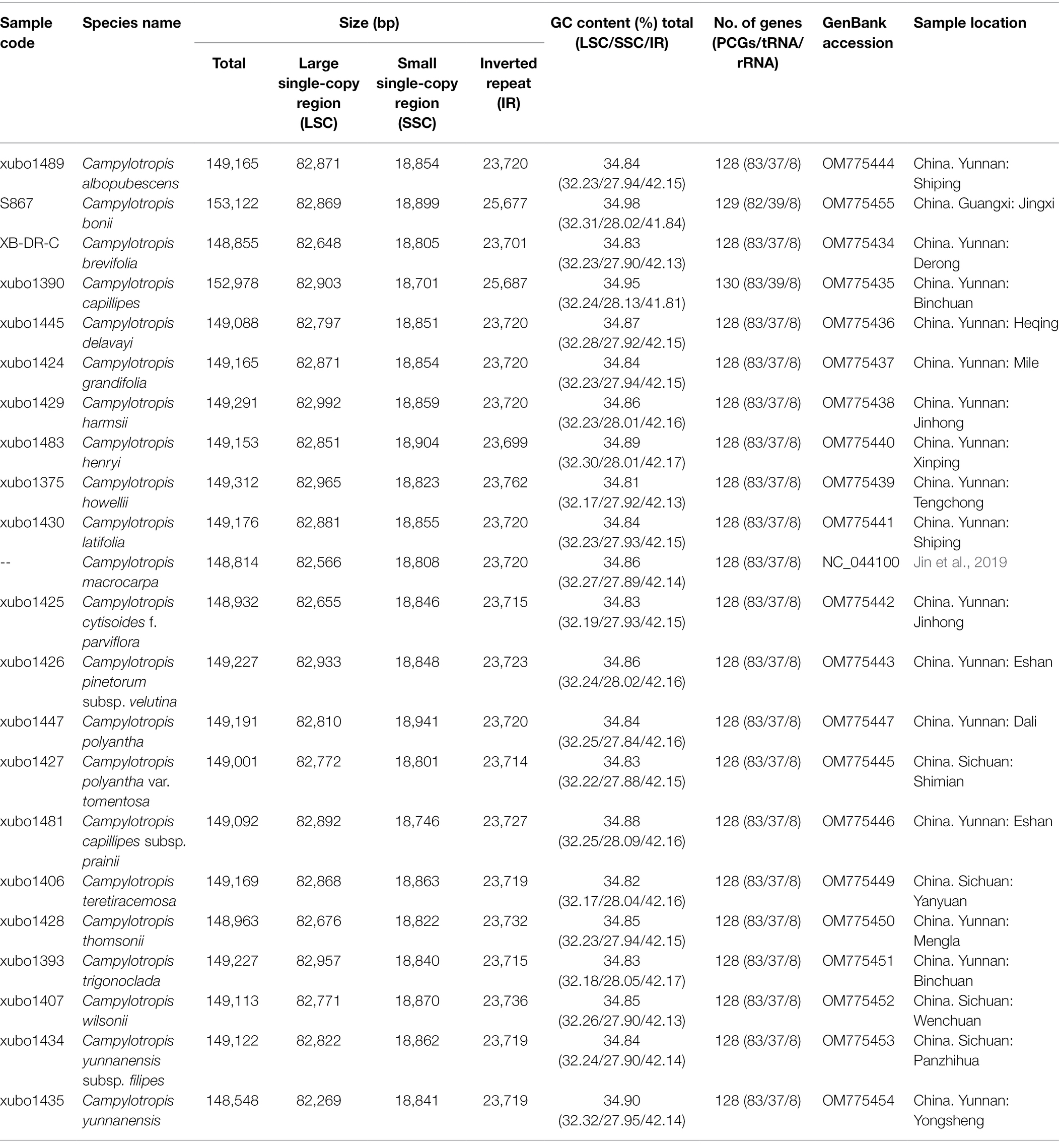
Table 1. Characteristics of the 22 complete chloroplast genomes for Campylotropis, including 21 newly generated accessions and the previously published accession of Campylotropis macrocarpa.
Chloroplast Genome Assembly, Annotation, and Comparison
For each accession, ~25 Gb of raw data were generated with pair-end 150 bp read length. Trimmomatic v0.39 (Bolger et al., 2014) was used to remove low-quality and adapter-containing reads. The clean data were then assembled using GetOrganelle v1.7.5 (Jin et al., 2020). Plastid Genome Annotator (Qu et al., 2019) was used to annotate the cp genomes based on one published accession of Campylotropis (C. macrocarpa; NC_044100; Jin et al., 2019) and 15 accessions of closely related legume species (Supplementary Table S2). Manual corrections for start and stop codons and the determination of pseudogenes were performed in Geneious v11 (Biomatters Ltd., Auckland, New Zealand). For the matK pseudogene annotated in the cp genome of C. bonii (see section “Results”), we further mapped raw reads to the assembled sequence of the matK gene, and performed Sanger sequencing to validate the accuracy of the assembled sequence. Raw reads were remapped to 400-bp surroundings of the IRb ends to quantify the IR junctions. Genome map of the cp genomes was generated using the online OrganellarGenome DRAW tool (OGDRAW; Lohse et al., 2013). To compare the contraction and expansion of IRs among cp genomes of Campylotropis and closely related genera, we identified and visualized boundaries of LSC, SSC, and IRs of the 25 whole cp genomes (including 22 Campylotropis accessions, two Lespedeza accessions, and Kummerowia striata) using IRscope (Amiryousefi et al., 2018).
Repeat Sequence Analysis
For 21 newly generated cp genomes and the published accession of C. macrocarpa, SSRs were identified using MISA software (Beier et al., 2017) with parameter settings of 11 for mono-, 6 for di-, 5 for tri-, 4 for tetra-, and 3 for penta- and hexa-nucleotide SSRs. For each of the 22 Campylotropis cp genomes, forward, reverse, palindrome, and complementary repeat sequences in LSC, IRb, and SSC regions were identified using REPuter program (Kurtz et al., 2001).
Molecular Marker Identification
The 22 whole cp genomes were firstly aligned using MAFFT v7 (Katoh and Standley, 2013). To identify hypervariable regions that can be used in species identification and phylogenetic studies for Campylotropis, nucleotide diversity (Pi) values were calculated in sliding windows along the alignment with a window length of 600 bp and step size of 200 bp. Pi values of each window were calculated using a custom Python script,1 with the formula referring to the algorithm implemented in pixy (Korunes and Samuk, 2021) to obtain unbiased estimations of nucleotide diversity in the presence of alignment gaps. Adjacent windows with a Pi value > 0.01 and a number of parsimony informative sites >25 were joined together as one single hypervariable region. The number of singleton variable sites, number of parsimony informative sites, and Pi values were calculated for each hypervariable region using the custom Python script.
Phylogenetic Analysis
To estimate the cp-genome-based phylogenetic relationships of Campylotropis as well as the tribe Desmodieae, we included the whole cp genomes of 22 Campylotropis accessions and 15 outgroups (Supplementary Table S2). The phylogenetic analyses were performed using Maximum likelihoods (ML) and Bayesian inference (BI) methods based on both whole cp genomes and shared protein-coding genes (PCGs). For the former dataset, MAFFT v7 was used to obtain the alignment of 37 whole cp genomes. As for the latter dataset, the shared PCGs were extracted and translated into amino acid sequences, and ClustalW2 (Larkin et al., 2007) was used to align the amino acid sequences. The codon alignment of each PCGs was obtained using PAL2NAL (Suyama et al., 2006). The ML trees were inferred using RAxML v8 (Stamatakis, 2014) based on the alignment of 37 whole cp genomes and the concatenated matrix of 72 PCGs. For each RAxML analysis, GTRGAMMA + I was set as the nucleotide substitution model and 1,000 bootstrap replicates were conducted to determine branch support. The BI analyses were performed using MrBayes v3.2 (Ronquist et al., 2012) with the nucleotide substitution model GTR + G + I (lset nst = 6 rates = invgamma). For each analysis, the posterior probability was estimated with two independent Markov Chain Monte Carlo (MCMC) chains (10 million generations and sampled every 1,000 generations) with the preliminary 25% of sampled data discarded as burn-in.
Analysis of Selective Pressure
To explore the selective pressure of PCGs in Campylotropis, the CODEML program implemented in the PAML v4.9 package (Yang, 2007) was used to estimate the rate of non-synonymous (dN) and synonymous (dS) substitutions for PCGs. In general, the ratio of dN/dS (ω) was supposed to equal 1 when under neutral evolution, a larger ω indicates higher positive selection pressure, while a smaller ratio of ω indicates higher pressure of negative selection.
All the 37 accessions in the above phylogenetic analysis were included, and the resulting phylogenetic tree was used as the input topology for CODEML. The codon-wise alignments of nucleotide sequences, which were used as the input sequences for CODEML, were generated with PAL2NAL (Suyama et al., 2006) guided by the peptide alignments. To determine whether each shared PCG has undergone a different evolutionary force in different lineages, we ran branch-site models with a one-ratio model (null hypothesis; ω0) in which all branches share the same ω and a two-ratio model in which the foreground branches (Campylotropis spp.; ωf) have a different ω (alternative hypothesis; ωb). Likelihood ratio tests with χ2 distribution were used to determine whether the alternative hypothesis significantly differ from the null hypothesis (Chi-square test, p < 0.05).
Results
Characteristics of Campylotropis cp Genomes
In this study, a total of 21 whole cp genomes of Campylotropis were newly generated and were submitted to GenBank under the accession numbers list in Table 1. Taken together with the previously published one of C. macrocarpa (NC_044100), the whole cp genomes of Campylotropis ranged from 148,548 bp (C. yunnanensis) to 153,122 bp (C. bonii), exhibiting a typical quadripartite structure comprising two IR regions (IRa and IRb) of 23,699–25,687 bp, an LSC region of 82,269–82,992 bp, and an SSC region of 18,746–18,941 bp (Table 1). The GC contents of the Campylotropis cp genomes were similar (34.81%–34.93%; Table 1). The IRs have the highest GC content (41.81%–42.18%), followed by the LSC region (32.17%–32.32%), and the SSC region (27.84%–28.13%).
The Campylotropis cp genomes were similar in gene contents, most of which encode 128 genes, including 83 PCGs, 37 tRNA genes, and eight rRNA genes (all located in the IRs; Table 1; Figure 1). Three species had a few pseudogenes and/or duplicated genes (Table 2). Specifically, C. capillipes and C. bonii has two more copies of the trnI-CAU gene, and C. bonii has a pseudogene (ψmatK; Table 2), which was confirmed by both raw reads mapping and Sanger sequencing (see Supplementary Figure S1 and Supplementary Dataset). Among the 83 PCGs, 77 were unique, and six (ndhB, rpl12, rpl23, rps7, rps12, and ycf2) were duplicated due to their location in the IRs. Likewise, 30 of the tRNA genes are unique, while seven tRNA genes (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC) and all four rRNA genes (rrn23, rrn16, rrn5, and rrn4.5) were duplicated. Eight PCGs (petB, petD, atpF, ndhB, ndhA, rpoC1, rpl16, and rps16) and six tRNA genes (trnA-UGC, trnI-GAU, trnG-UCC, trnL-UAA, trnV-UAC, trnK-UUU) contained one intron, while only three PCGs (rps12, ycf3, and clpP) contained two introns (Table 2). In all newly generated Campylotropis cp genomes, the 5′ end of the rps12 gene was located in the LSC region, and the 3′ end was duplicated in the IRs.
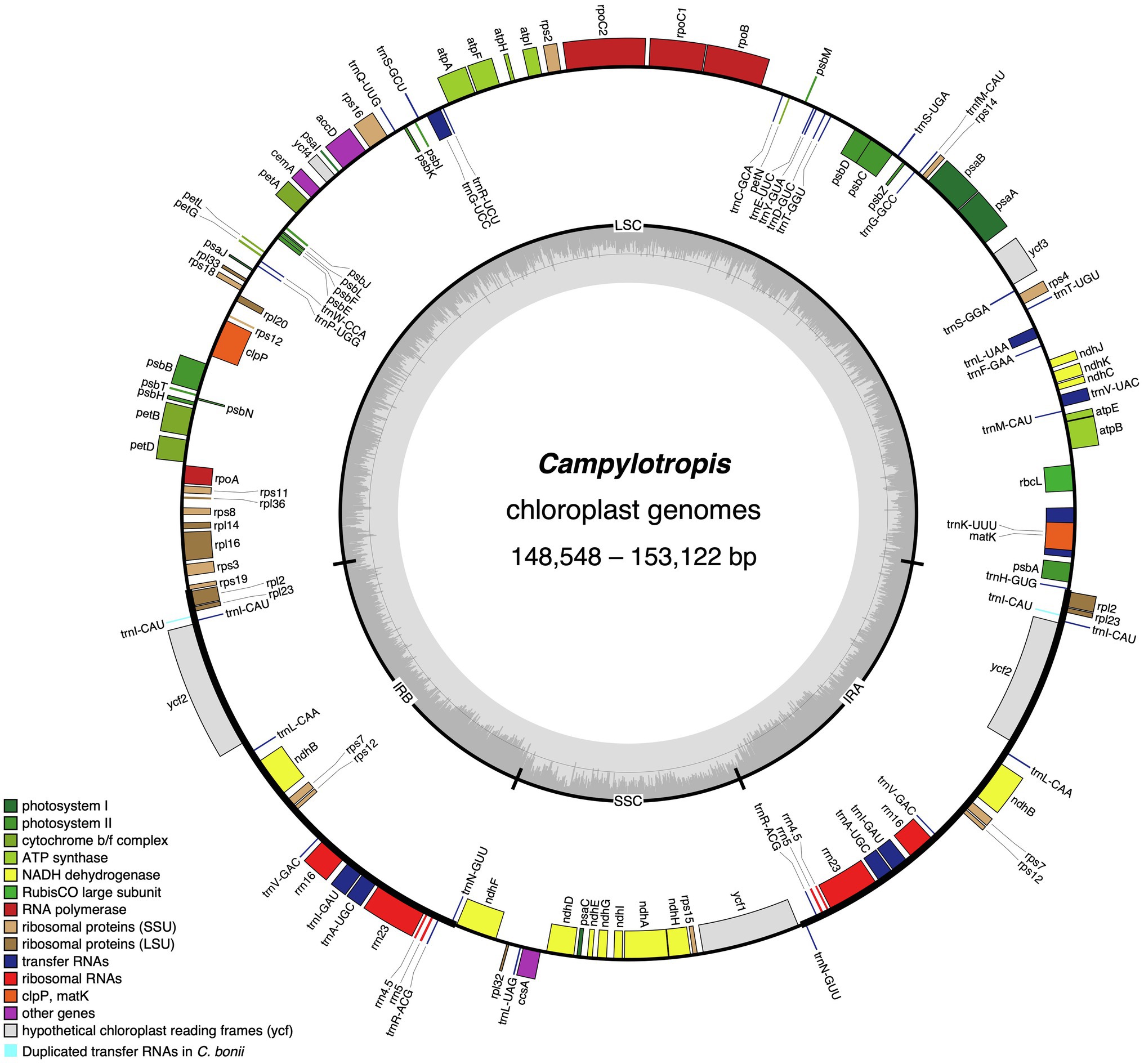
Figure 1. The chloroplast genome map of Campylotropis species. Genes inside and outside of the circle are transcribed clockwise and counterclockwise, respectively. Genes belonging to different functional groups are shown in different colors, with extra duplicated genes in Campylotropis bonii highlighted in light blue. The dark gray area in the inner circle denotes GC content while the light gray corresponds to the AT content of the genome. LSC, large single copy; SSC, small single copy; and IR, inverted repeat.

Table 2. Summary of gene contents present in the Campylotropis chloroplast genomes.
Comparative Analysis of IR Boundaries
The IR boundary of the assembled cp genomes were quantified by the remapping of short reads, which showed above 300× for the IRb ends and surrounding areas (Supplementary Table S3). We compared the IR boundaries of 25 cp genomes from subtribe Lespedezinae, including Lespedeza maritima, Lespedeza cuneata, Kummerowia striata, and 22 Campylotropis accessions, and found a little variation of the expansion/contraction of the IRs (Supplementary Figure S2). The JLA (IRa-LSC) and JSA (IRa-SSC) boundaries are highly consistent in the 25 cp genomes, with the former located between rpl2 and trnH, and the latter between ycf1 and trnN. The distances between the JLA boundary and trnH were 0–19 bp, while those between the JSA boundary and ycf1 varied from 128 to 144 bp. The JLB (IRb-LSC) boundaries cut through rps19 in most species, with 32–48 bp of rps19 extended into the IRb, while the JLB boundaries of C. thomsonii and C. parviflora were 88 bp away from rps19 due to the contraction of IRs. The distance between the JSB (IRb-SSC) boundaries and ndhF varied from 2 to 33 bp in most species except Kummerowia striata, where ndhF extended 11 bp into IRb due to the expansion of IRs.
Characteristics of Repeat Sequences
The number of SSRs in the Campylotropis species varied from 50 in C. harmsii to 115 in C. teretiracemosa (Supplementary Table S4), in which mononucleotide SSRs were most abundant, followed by component and dinucleotides SSRs (Figure 2A). Among the motifs in the SSRs, A/T, AA/TT, and AT/AT were the most frequently occurring motifs (Figure 2B). Besides, most of the SSRs were located in the LSC (38–56) and SSC (10–18) regions, and very few were located in the IRs (Supplementary Table S5). REPuter identified 40–71 repeat sequences with length > 30 bp, covering 1,647–4,278 bp in the cp genomes of Campylotropis species (Figures 2C,D). Palindromic repeat sequences were most abundant (22–32), followed by forward (14–20) and reverse (2–13) repeat sequences (Figure 2C; Supplementary Table S6). All the repeat sequences with length > 30 bp were located in LSC (33–64) and IRs (6–10), while none of them were identified in the SSC region (Supplementary Table S6). Most of the repeat sequences were less than 100 bp, a few of them were larger than 100 bp (Figure 2D; Supplementary Table S6). Notably, C. bonii and C. capillipes each had a forward repeat sequence with a length of 2,219 and 2,217 bp, respectively (Figure 2D; Supplementary Table S7). Both repeat sequences were located between rpl23 and ycf2 in the IRs, which caused the duplication of trnI-CAU and resulted in four copies of this gene (Supplementary Figure S3).
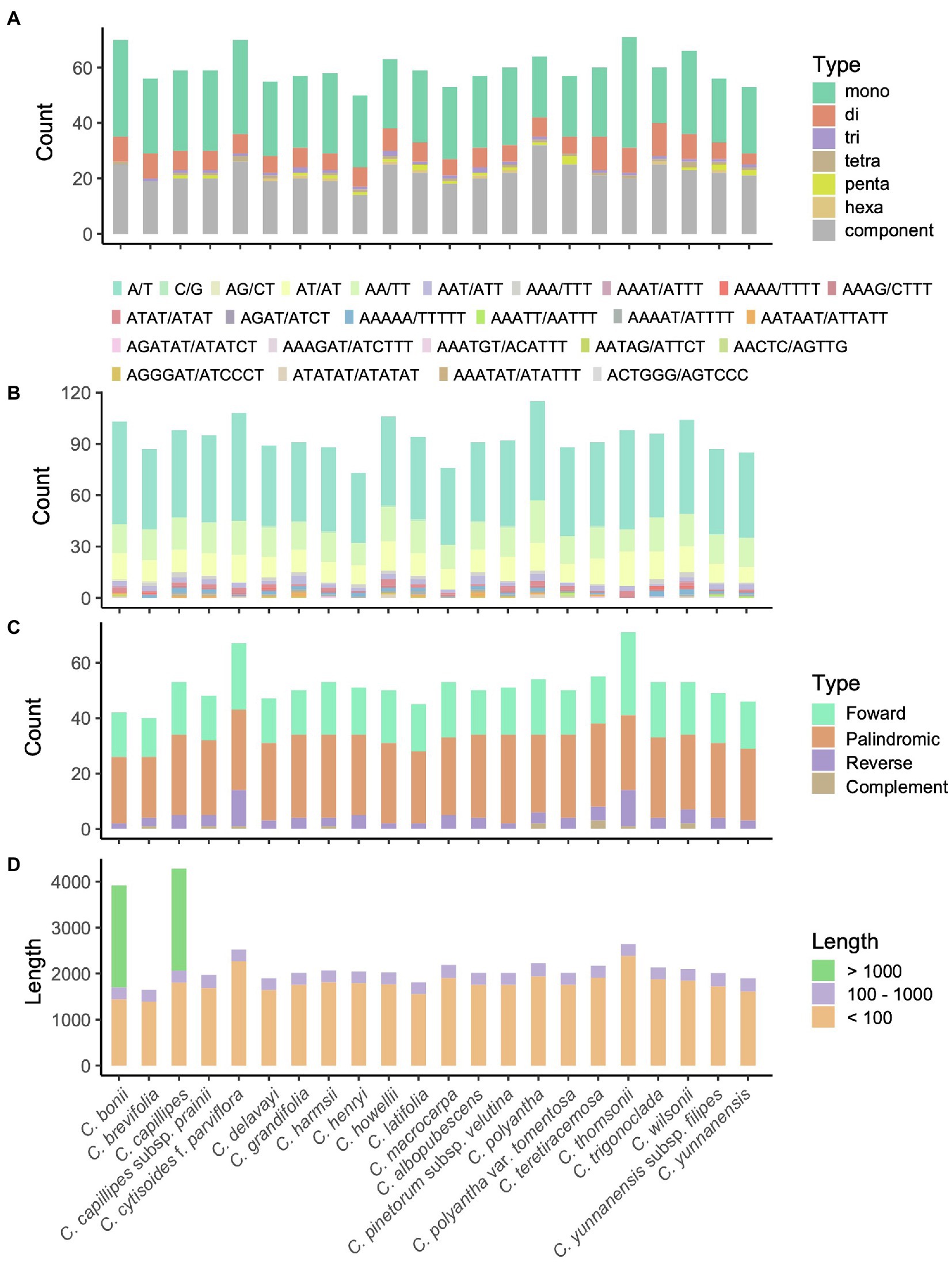
Figure 2. Patterns of simple sequence repeats (SSRs; A,B) and long sequence repeats (LSRs; C,D) for the 23 chloroplast genomes of Campylotropis species. (A) Number of motifs and their abundance of SSRs in each species. (B) Type of motifs and their abundance of SSRs in each species. (C) Type and abundance of LSRs in each species. (D) Accumulative length of LSRs in each species.
Identification of Candidate Molecular Markers
Using sliding window analysis, we found that most genetic variations in the cp genomes of Campylotropis occurred in the LSC and SSC regions (Figure 3). A total of 13 intergenic spacer regions located in the LSC region, ranging from 547 to 1,995 bp, were identified as potential molecular markers for phylogenetic and population genetic studies (Figure 3; Table 3). Among them, the intergenic spacer of atpA and psbI (atpA-psbI) was the longest (1,995) and contained the greatest number of parsimony informative sites (109), while the intergenic spacer of ycf4 and cemA (ycf4-cemA) had the highest Pi value (0.0117).
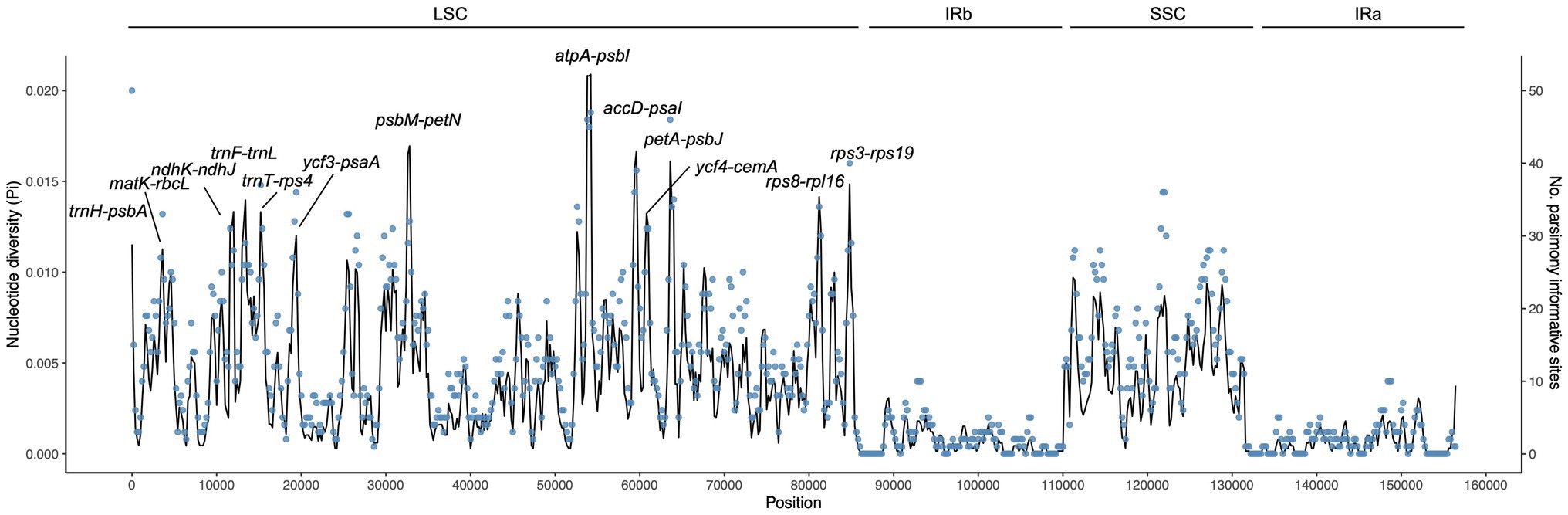
Figure 3. Nucleotide diversity (Pi, black line, vertical left axis) and number of parsimony informative sites (blue dots, vertical right axis) of the Campylotropis chloroplast genomes based on sliding window analysis. The window length is 600 bp and the step size is 200 bp. The horizontal axis indicates the position of the midpoint of a window. The 13 regions with high diversity are indicated above the peaks.
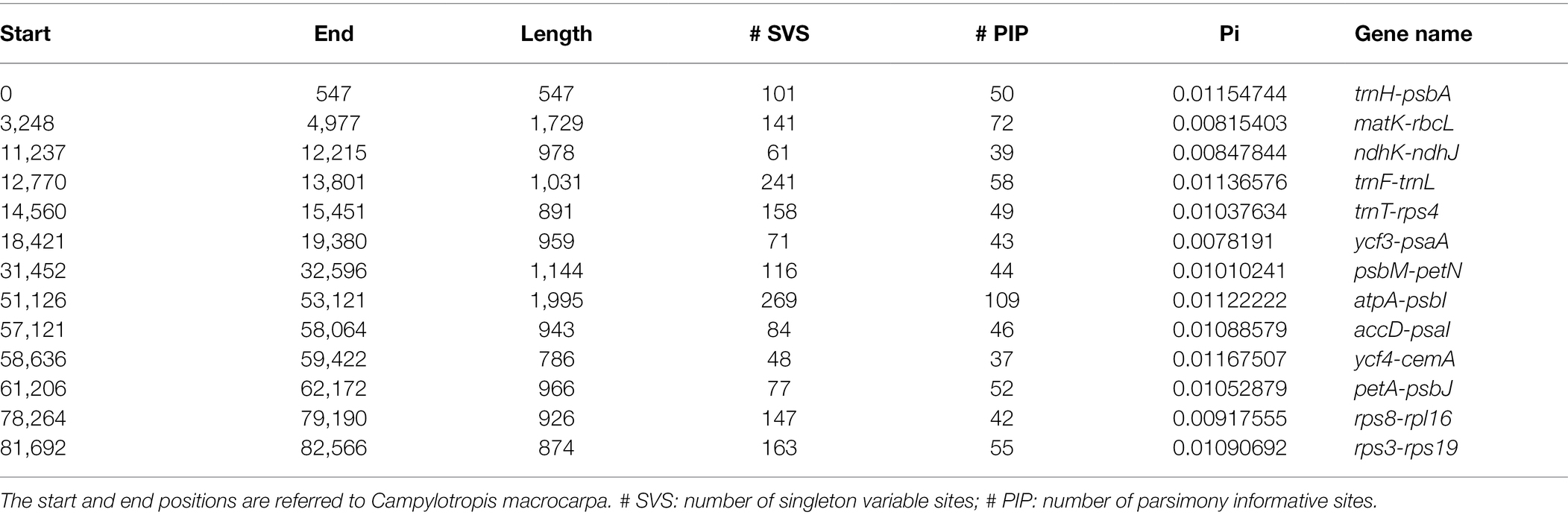
Table 3. Hypervariable regions identified among the 22 cp genomes of Campylotropis.
Phylogenetic Relationships of Campylotropis
The phylogenetic trees inferred from Maximum likelihood (ML) and Bayesian inference (BI) based on the whole cp genome shared an identical topology and showed little differences in support values (Figure 4). The concatenated alignment of PCGs resulted in similar topologies, with a few differences with regard to the relationships within Campylotropis (Supplementary Figures S4, S5). All topologies fully supported the reciprocal monophyly of the two subtribes in tribe Desmodieae [100% bootstrap support (BS) and 1 posterior probability (PP)]. In the subtribe Lespedezinae, Kummerowia striata and the two Lespedeza species formed a clade (BS = 100%, PP = 1), and Campylotropis was also a monophyletic clade (BS = 100%, PP = 1).
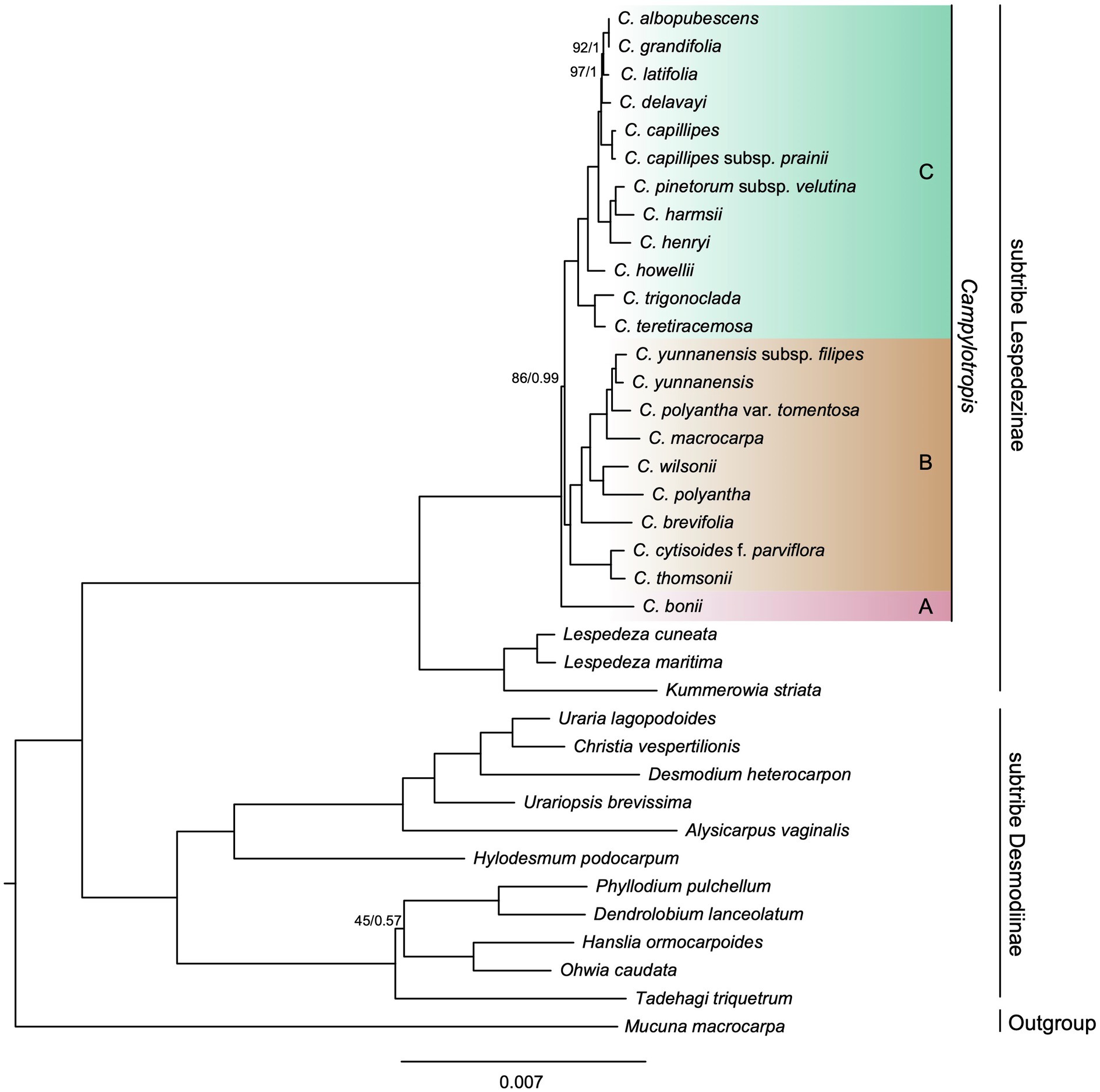
Figure 4. Phylogenetic tree obtained using the Maximum Likelihood (ML) and Bayesian Inference (BI) method for Campylotropis spp. and closely related species based on whole cp genomes. Numbers above branches indicate ML bootstrap supports (BS; before the slash) and Bayesian posterior probabilities (PP; after the slash). The full support values are not indicated.
As for the relationship within Campylotropis, both ML and BI trees based on the whole cp genome supported C. bonii (lineage A) as sister to the remaining species (Figure 4), and the latter clade (BS = 86%, PP = 0.99) segregated into two subclades (lineages B and C), each with full support values (BS = 100%, PP = 1). Lineage B included C. yunnanensis subsp. filipes, C. yunnanensis, C. polyantha var. tomentosa, C. macrocarpa, C. wilsonii, C. polyantha, C. brevifolia, C. cytisoides f. parviflora, and C. thomsonii. And, lineage C included C. albopubescens, C. grandifolia, C. latifolia, C. delavayi, C. capillipes, C. capillipes subsp. prainii, C. pinetorum subsp. velutina, C. harmsii, C. henryi, C. howellii, C. trigonoclada, and C. teretiracemosa. The ML three based on the PCGs dataset showed the same topology as that based on the whole cp genome with regard to the relationship among the three subclades of Campylotropis, albeit the supporting values were lower (lineage B: BS = 93%; lineage B sister to lineage C: BS = 79%; Supplementary Figure S4). However, the BI inference based on the PCGs dataset revealed a different topology, in which C. bonii was weakly supported to be a sister clade of lineage B (PP = 0.604; Supplementary Figure S5).
Selective Pressure of cp Genes in Campylotropis
A total of 68 shared PCGs were subjected to the selective pressure analysis (Supplementary Table S8). Most of the genes were subjected to purifying selection (ω < 1; Figure 5). Using the likelihood ratio test, we found that 11 genes showed significantly different selective pressure in Campylotropis (Figure 5; Supplementary Table S8). Among them, two genes (atpF and rps19) showed obvious signatures of positive selection (ωf > 1, p < 0.05) in Campylotropis and eight genes (ndhC, ndhD, psbA, rpoC1, rpoC2, rps4, ycf1, and ycf2) evolved faster in Campylotropis than in the background branches (ωf > ωb, p < 0.05; Figure 5; Supplementary Table S8).
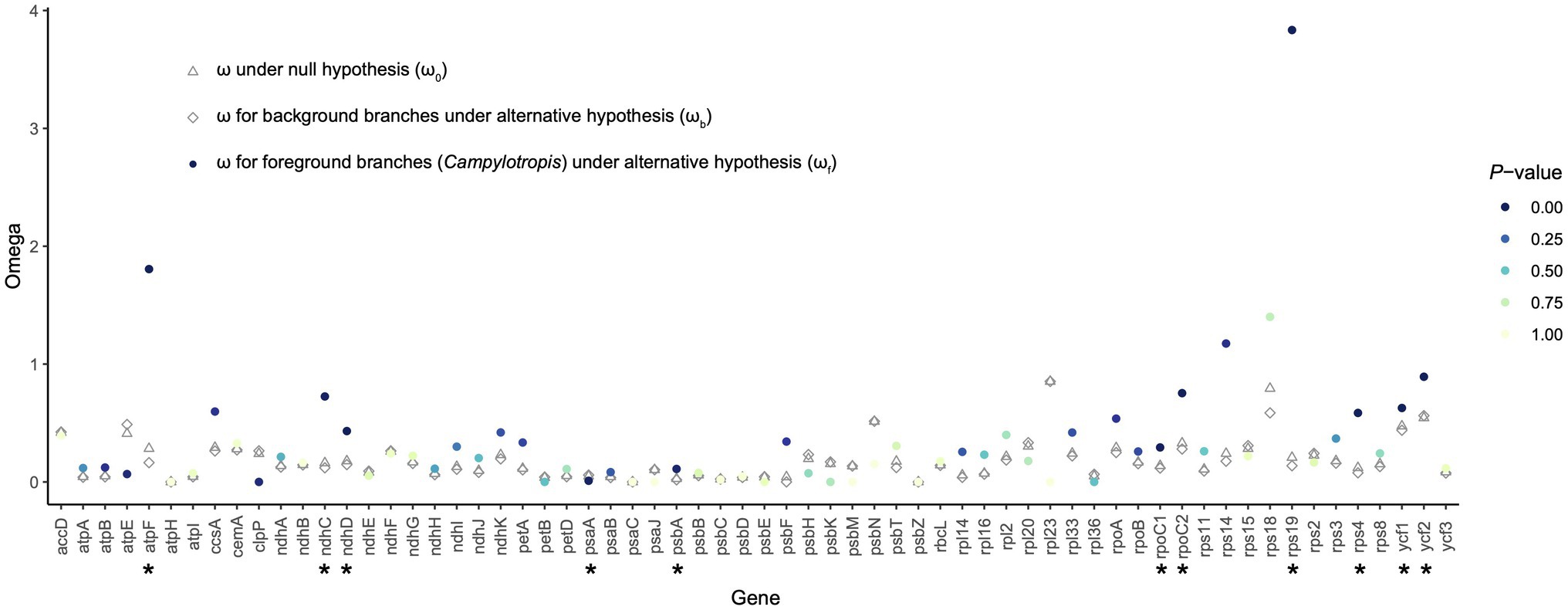
Figure 5. The ratio of non-synonymous (dN) and synonymous (dS) substitutions (dN/dS; ω) for protein-coding genes shared by the Desmodieae species. The asterisks under the gene names indicate statistical significance (p < 0.05) between the null hypothesis and the alternative hypothesis.
Discussion
Variations and Evolution of Whole cp Genomes in Campylotropis
The 21 newly assembled and one previously published Campylotropis cp genomes showed little variation in genome structure and genome length, as found in other legume species (Wang et al., 2018; Oyebanji et al., 2020; Zhang et al., 2020; Liao et al., 2021). The Campylotropis cp genomes exhibit the typical quadripartite structure and no large structural variant was found (Table 1). The genome length of these species was similar (148,548–153,122 bp) and fell within the range of subfamily Papilionoideae (c. 140–160 kb; Oyebanji et al., 2020). Other genome features, including lengths of LSC, SSC, and IRs, expansion and contraction of IR boundaries, number of genes, GC content, the pattern of SSRs also varied little within this genus, which is comparable to other genera from the legume family (e.g., Oyebanji et al., 2020; Liao et al., 2021).
Despite the general homogeneity characteristics mentioned above, there are some interesting inconsistencies worth mentioning in Campylotropis cp genomes. Previous studies demonstrated that expansion and contraction of IRs substantially contribute to the change in the size of cp genomes (Ruhlman and Jansen, 2014; Zheng et al., 2017; Gu et al., 2020). In our study, the JLB (IRb-LSC) boundaries cut through rps19 in most species, except in C. thomsonii and C. parviflora, where JLB was located between rps19 and rpl2, causing less than 100-bp length variation of the IRs (Supplementary Figure S2). However, the cp genomes of C. bonii and C. capillipes were 3–4 kb longer than the rest without showing any significant signal of IR expansion (Figure 2; Supplementary Figure S2). Both cp genomes have a ~2 kb long sequence repeat in each IR region, causing a ~4 kb increase in total genome length. These results indicate that similar to nuclear genomes (Bennetzen et al., 2005), dynamics in repeat sequences rather than expansion and contraction of IRs played an important role in the length variation of Campylotropis cp genomes. The long sequence repeats also caused duplication of trnI-CAU and resulted in four copies of this gene (Supplementary Figure S1; Supplementary Table S7).
The Campylotropis cp genomes showed moderate sequence variation, most occurring in the LSC region (Figure 3). Consequently, all 13 candidate molecular markers were located in the LSC region, which may be useful in further studies of species delimitation, phylogenetic, and population genetic studies (Table 3). Many of these molecular markers have been reported in other studies, such as trnH-psbA (Li et al., 2021), accD-psaI (Chen et al., 2021), and petN-trnD (Liao et al., 2021). Notably, the matK gene, which encodes a protein essential for in vivo splicing of Group II introns (Ahlert et al., 2006), is a pseudogene in C. bonii. As one of the most frequently used molecular markers in angiosperm phylogenetic studies (Patwardhan et al., 2014), matK has a high overall evolutionary rate in contrast to other chloroplast genes (Wanke et al., 2007). In fact, pseudogenic copies of matK pseudogene were reported in orchids (Kocyan et al., 2008), Piperales (Wanke et al., 2007), and Ericaceae (Braukmann et al., 2017). In the legume family, matK pseudogenes were found in Tadehagi triquetrum (GenBank accession: MW557314.1; unpublished) and reported in Tylosema spp. (Wang et al., 2018). Here, C. bonii provided one more case for legume plants living with pseudogenic matK gene.
Phylogenetic Relationships
The phylogenetic trees reconstructed on both whole cp genome and shared PCGs in this study fully supported the monophyly of the two subtribes of Desmodieae (Figure 4; Supplementary Figures S4, S5). The subtribe Desmodiinae was divided into two fully supported monophyletic groups as described in previous studies (Jabbour et al., 2018; Jin et al., 2019). Subtribe Lespedezinae consist of three genera: Campylotropis, Lespedeza, and Kummerowia (Figure 4). Since the first Chinese species of Campylotropis (C. macrocarpa) was described as Lespedeza macrocarpa Bunge (Bunge, 1835), a number of species have been recorded under Lespedeza, Campylotropis was thought to be derived from Lespedeza (Fu, 1987). However, molecular phylogenetic studies based on one or several molecular markers found a sister relationship between Lespedeza and Kummerowia (Xu et al., 2012; Jabbour et al., 2018). Likewise, whole cp genomes in both Jin et al. (2019) and this study confirmed that Lespedeza was sister to Kummerowia, and the two genera were then sister to Campylotropis.
The intrageneric and interspecific relationships of Campylotropis have been unsettled for a long time due to complex morphological characteristics and lack of molecular phylogenetic studies (e.g., Jabbour et al., 2018). Our results strongly support Campylotropis as a monophyletic group, consisting of three lineages (i.e., A, B, and C; Figure 4). Lineage A contains only one species, C. bonii, which was sister to all the remaining species of Campylotropis (lineage B and lineage C). Species from lineage C were mostly restricted in southwestern China and Southeast Asia, while lineage B contained regional endemic and widely distributed species. For example, among species in lineage B, C. wilsonii is endemic to western Sichuan while C. macrocarpa is distributed throughout southwestern China and East Asia (Huang et al., 2010). However, the relationships among the three lineages were not resolved, as the support value of the sister relationship between lineage B and lineage C was relatively low (Figure 4; Supplementary Figure S4), and the BI inference resulted in a different topology (Supplementary Figure S5). The former topology agrees with a previous study that included five Campylotropis species in the phylogenetic analysis of the tribe Desmodieae, but the results were only based on several molecular markers: chloroplast (rbcL, psbA-trnH) and nuclear (ITS-1) DNA sequences (Jabbour et al., 2018). Thus, phylogenetic studies with more extensive sampling and nuclear genomic data are needed to elucidate the intrageneric relationships of Campylotropis.
Selective Pressure
Positive selection is assumed to play key parts in the adaptation of organisms to diverse environments (Moseley et al., 2018), while negative (purifying) selection is a ubiquitous evolutionary force responsible for genomic sequence conservation across long evolutionary timescales (Cvijović et al., 2018). For example, the positive selection pressure of genes related to photosynthesis was found less than other types of genes (Du et al., 2016; Gao et al., 2018; Li et al., 2020). As expected, the ω values for most genes, especially photosynthesis genes, were less than 1, either in Campylotropis or in background branches (Figure 5). The two genes under significant positive selection in Campylotropis: atpF and rps19 (ωf > 1; p < 0.05) were also found under positive selection in other species, e.g., atpF in two deciduous Quercus species (Yin et al., 2018), and rps19 in Garcinia paucinervis (Wang et al., 2021). As indicated in Yin et al. (2018), atpF gene is highly divergent between deciduous and evergreen sclerophyllous oaks since the former loses its leaves in cold and drought seasons. Despite having ωf < 1, eight genes (ndhC, ndhD, psbA, rpoC1, rpoC2, rps4, ycf1, and ycf2) significantly accelerated their evolution in Campylotropis compared to background branches (ωf > ωb, p < 0.05). Some of them were reported to be under significant positive selection in other taxa, such as ycf1 in seed plants (Zheng et al., 2017), ndhC in Echinacanthus (Gao et al., 2019), and rpoC2 in Rehmannia (Zeng et al., 2017). Therefore, these positively selected and fast-evolving genes may play an important role in the adaptation of Campylotropis species to arid soils and various types of habitats.
Conclusion
In this study, we assembled 21 whole cp genomes for Campylotropis spp. Comparative analysis of the cp genome size, structure, expansion and contraction of IR boundaries, number of genes, GC content, and pattern of SSRs revealed high similarities among the Campylotropis cp genomes. The activities of long sequence repeats contributed to the variation in genome size and gene content in Campylotropis cp genomes. The Campylotropis cp genomes showed moderate sequence variation, and 13 candidate regions were identified for further studies of species identification and phylogenetic studies. We also reported one more case of matK pseudogene for legume species in C. bonii. The phylogenetic analysis confirmed the monophyly of Campylotropis and the sister relationship between Lespedeza and Kummerowia, the latter two genera were then sister to Campylotropis. And, its intrageneric relationships based on genomic scale data were firstly reported in this study. The two positively selected genes (atpF and rps19) and eight fast-evolving genes identified in this study may help us to understand the adaptation of Campylotropis species.
Data Availability Statement
The sequences and annotations of the newly generated chloroplast genomes of Campylotropis species were deposited in the National Center for Biotechnology Information (NCBI) GenBank database under the accession numbers list in Table 1.
Author Contributions
YF, X-FG, and BX conceived and designed the study. BX, H-ND, J-YZ, and ML collected the sample. YF, J-YZ, L-SJ, and XL analyzed the data. YF wrote the manuscript. BX revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 31570196), the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (grant no. 2019QZKK0502), and Wild Plants Sharing and Service Platform of Sichuan Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Shi-Qi Li and Xue-Ping Fan for their help with sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.895543/full#supplementary-material
Footnotes
References
Ahlert, D., Piepenburg, K., Kudla, J., and Bock, R. (2006). Evolutionary origin of a plant mitochondrial group II intron from a reverse transcriptase/maturase-encoding ancestor. J. Plant Res. 119, 363–371. doi: 10.1007/s10265-006-0284-0
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Barham, J. (1997). 327. Campylotropis polyantha: Leguminosae-Papilionoideae. Curtis’s Bot. Mag. 203–207.
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bennetzen, J. L., Ma, J., and Devos, K. M. (2005). Mechanisms of recent genome size variation in flowering plants. Ann. Bot. 95, 127–132. doi: 10.1093/aob/mci008
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Braukmann, T. W. A., Broe, M. B., Stefanović, S., and Freudenstein, J. V. (2017). On the brink: the highly reduced plastomes of nonphotosynthetic Ericaceae. New Phytol. 216, 254–266. doi: 10.1111/nph.14681
Bunge, A. (1835). Plantarum mongholico-chinensium decas prima. Uchenya Zap. Imp. Kazan. Univ. 4, 154–180.
Cai, J., Ma, P. F., Li, H. T., and Li, D. Z. (2015). Complete plastid genome sequencing of four Tilia species (Malvaceae): a comparative analysis and phylogenetic implications. PLoS One 10:e0142705. doi: 10.1371/journal.pone.0142705
Chen, J., Zang, Y., Shang, S., Liang, S., Zhu, M., Wang, Y., et al. (2021). Comparative chloroplast genomes of Zosteraceae species provide adaptive evolution insights into seagrass. Front. Plant Sci. 12:741152. doi: 10.3389/fpls.2021.741152
Cvijović, I., Good, B. H., and Desai, M. M. (2018). The effect of strong purifying selection on genetic diversity. Genetics 209, 1235–1278. doi: 10.1534/genetics.118.301058
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Dopp, I. J., Yang, X., and Mackenzie, S. A. (2021). A new take on organelle-mediated stress sensing in plants. New Phytol. 230, 2148–2153. doi: 10.1111/nph.17333
Du, Q., Bi, G., Mao, Y., and Sui, Z. (2016). The complete chloroplast genome of Gracilariopsis lemaneiformis (Rhodophyta) gives new insight into the evolution of family Gracilariaceae. J. Phycol. 52, 441–450. doi: 10.1111/jpy.12406
Gao, C., Deng, Y., and Wang, J. (2019). The complete chloroplast genomes of Echinacanthus species (Acanthaceae): phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front. Plant Sci. 9:1989. doi: 10.3389/fpls.2018.01989
Gao, X., Zhang, X., Meng, H., Li, J., Zhang, D., and Liu, C. (2018). Comparative chloroplast genomes of Paris sect. Marmorata: insights into repeat regions and evolutionary implications. BMC Genomics 19, 133–144. doi: 10.1186/s12864-018-5281-x
Gu, L., Su, T., An, M. T., and Hu, G. X. (2020). The complete chloroplast genome of the vulnerable Oreocharis esquirolii (Gesneriaceae): structural features, comparative and phylogenetic analysis. Plan. Theory 9, 1–16. doi: 10.3390/plants9121692
Han, B., Jiang, P., Liu, W., Xu, H., Li, Y., Li, Z., et al. (2018). Role of daucosterol linoleate on breast cancer: studies on apoptosis and metastasis. J. Agric. Food Chem. 66, 6031–6041. doi: 10.1021/acs.jafc.8b01387
Howe, C. J., Barbrook, A. C., Koumandou, V. L., Nisbet, R. E. R., Symington, H. A., Wightman, T. F., et al. (2003). Evolution of the chloroplast genome. Philos. Trans. R. Soc. B Biol. Sci. 358, 99–107. doi: 10.1098/rstb.2002.1176
Huang, P., Ohashi, H., Iokawa, Y., and Nemoto, T. (2010). “Fabaceae, tribe desmodieae,” in Flora of China. eds. C. Y. Wu, P. H. Raven, and D. Y. Hong (St. Louis: Science Press, Beijing, and Missouri Botanical Garden), 262–311.
Iokawa, Y., and Ohashi, H. (2008). Campylotropis (Leguminosae) of China, an enumeration and distribution. J. Japanese Bot. 83, 36–59.
Jabbour, F., Gaudeul, M., Lambourdière, J., Ramstein, G., Hassanin, A., Labat, J. N., et al. (2018). Phylogeny, biogeography and character evolution in the tribe Desmodieae (Fabaceae: Papilionoideae), with special emphasis on the new Caledonian endemic genera. Mol. Phylogenet. Evol. 118, 108–121. doi: 10.1016/j.ympev.2017.09.017
Jin, D. P., Choi, I. S., and Choi, B. H. (2019). Plastid genome evolution in tribe Desmodieae (Fabaceae: Papilionoideae). PLoS One 14, 1–17. doi: 10.1371/journal.pone.0218743
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Depamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keller, J., Rousseau-Gueutin, M., Martin, G. E., Morice, J., Boutte, J., Coissac, E., et al. (2017). The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res. 24, 343–358. doi: 10.1093/dnares/dsx0006
Kocyan, A., Vogel, E. F. D., Conti, E., and Gravendeel, B. (2008). Molecular phylogeny of Aerides (Orchidaceae) based on one nuclear and two plastid markers: A step forward in understanding the evolution of the Aeridinae. Mol. Phylogenet. Evol. 48, 422–443. doi: 10.1016/j.ympev.2008.02.017
Korunes, K. L., and Samuk, K. (2021). Pixy: unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol. Ecol. Resour. 21, 1359–1368. doi: 10.1111/1755-0998.13326
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Li, Y., Dong, Y., Liu, Y., Yu, X., Yang, M., and Huang, Y. (2021). Comparative analyses of euonymus chloroplast genomes: genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front. Plant Sci. 11:593984. doi: 10.3389/fpls.2020.593984
Li, C., Zhao, Y., Xu, Z., Yang, G., Peng, J., and Peng, X. (2020). Initial characterization of the chloroplast genome of Vicia sepium, an important wild resource plant, and related inferences about its evolution. Front. Genet. 11:73. doi: 10.3389/fgene.2020.00073
Liao, M., Gao, X.-F., Zhang, J.-Y., Deng, H.-N., and Xu, B. (2021). Comparative chloroplast genomics of Sophora species: evolution and phylogenetic relationships in the early-diverging legume subfamily Papilionoideae (Fabaceae). Front. Plant Sci. 12:778933. doi: 10.3389/fpls.2021.778933
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Lu, R. S., Li, P., and Qiu, Y. X. (2017). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7:2054. doi: 10.3389/fpls.2016.02054
Luo, Y., Ma, P. F., Li, H. T., Yang, J. B., Wang, H., and Li, D. Z. (2016). Plastid phylogenomic analyses resolve Tofieldiaceae as the root of the early diverging monocot order Alismatales. Genome Biol. Evol. 8, 932–945. doi: 10.1093/gbe/evv260
Moseley, R. C., Mewalal, R., Motta, F., Tuskan, G. A., Haase, S., and Yang, X. (2018). Conservation and diversification of circadian rhythmicity between a model crassulacean acid metabolism plant kalanchoë fedtschenkoi and a model C3 photosynthesis plant arabidopsis thaliana. Front. Plant Sci. 9:1757. doi: 10.3389/fpls.2018.01757
Oyebanji, O., Zhang, R., Chen, S. Y., and Yi, T. S. (2020). New insights into the plastome evolution of the Millettioid/Phaseoloid clade (Papilionoideae, Leguminosae). Front. Plant Sci. 11:151. doi: 10.3389/fpls.2020.00151
Patwardhan, A., Ray, S., and Roy, A. (2014). Molecular markers in phylogenetic studies—a review. J. Phylogenetics Evol. Biol. 2:131. doi: 10.4172/2329-9002.1000131
Qu, X. J., Moore, M. J., Li, D. Z., and Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 1–12. doi: 10.1186/s13007-019-0435-7
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Ruhlman, T. A., and Jansen, R. K. (2014). The plastid genomes of flowering plants. Methods Mol. Biol. 1132, 3–38. doi: 10.1007/978-1-62703-995-6_1
Ruhsam, M., Rai, H. S., Mathews, S., Ross, T. G., Graham, S. W., Raubeson, L. A., et al. (2015). Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in araucaria? Mol. Ecol. Resour. 15, 1067–1078. doi: 10.1111/1755-0998.12375
Sabir, J., Schwarz, E., Ellison, N., Zhang, J., Baeshen, N. A., Mutwakil, M., et al. (2014). Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat-lacking clade of legumes. Plant Biotechnol. J. 12, 743–754. doi: 10.1111/pbi.12179
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Wang, Y. H., Wicke, S., Wang, H., Jin, J. J., Chen, S. Y., Zhang, S. D., et al. (2018). Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae). Front. Plant Sci. 9:138. doi: 10.3389/fpls.2018.00138
Wang, Y., Zhao, B., Lu, Z., Shi, Y., and Li, J. (2021). The complete chloroplast genome provides insight into the polymorphism and adaptive evolution of Garcinia paucinervis. Biotechnol. Biotechnol. Equip. 35, 377–391. doi: 10.1080/13102818.2021.1879676
Wanke, S., Jaramillo, M. A., Borsch, T., Samain, M. S., Quandt, D., and Neinhuis, C. (2007). Evolution of Piperales—matK gene and trnK intron sequence data reveal lineage specific resolution contrast. Mol. Phylogenet. Evol. 42, 477–497. doi: 10.1016/j.ympev.2006.07.007
Wen, P., Han, H., Wang, R., Wang, N., and Yao, X. (2007). C-glycosylfavones and aromatic glycosides from Campylotropis hirtella (Franch.) Schindl. Asian J. Tradit. Med. 2, 149–153.
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Xu, B., Wu, N., Gao, X. F., and Zhang, L. B. (2012). Analysis of DNA sequences of six chloroplast and nuclear genes suggests incongruence, introgression, and incomplete lineage sorting in the evolution of lespedeza (Fabaceae). Mol. Phylogenet. Evol. 62, 346–358. doi: 10.1016/j.ympev.2011.10.007
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yin, K., Zhang, Y., Li, Y., and Du, F. K. (2018). Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. Int. J. Mol. Sci. 19:1042. doi: 10.3390/ijms19041042
Zeng, S., Zhou, T., Han, K., Yang, Y., Zhao, J., and Liu, Z. L. (2017). The complete chloroplast genome sequences of six Rehmannia species. Genes 8:103. doi: 10.3390/genes8030103
Zhang, S. D., Jin, J. J., Chen, S. Y., Chase, M. W., Soltis, D. E., Li, H. T., et al. (2017). Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zhang, R., Wang, Y. H., Jin, J. J., Stull, G. W., Bruneau, A., Cardoso, D., et al. (2020). Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 69, 613–622. doi: 10.1093/sysbio/syaa013
Zhang, L., Wang, S., Su, C., Harris, A. J., Zhao, L., Su, N., et al. (2021). Comparative chloroplast genomics and phylogenetic analysis of Zygophyllum (Zygophyllaceae) of China. Front. Plant Sci. 12:723622. doi: 10.3389/fpls.2021.723622
Zhao, C., Wang, Y., Chan, K. X., Marchant, D. B., Franks, P. J., Randall, D., et al. (2019). Evolution of chloroplast retrograde signaling facilitates green plant adaptation to land. Proc. Natl. Acad. Sci. U. S. A. 116, 5015–5020. doi: 10.1073/pnas.1812092116
Keywords: Campylotropis, legume, adaptive evolution, phylogenomics, comparative genomics, chloroplast genome
Citation: Feng Y, Gao X-F, Zhang J-Y, Jiang L-S, Li X, Deng H-N, Liao M and Xu B (2022) Complete Chloroplast Genomes Provide Insights Into Evolution and Phylogeny of Campylotropis (Fabaceae). Front. Plant Sci. 13:895543. doi: 10.3389/fpls.2022.895543
Edited by:
Ruslan Kalendar, University of Helsinki, FinlandReviewed by:
Revathi Sekar, Helmholtz Association of German Research Centres (HZ), GermanyM. Gonzalo Claros, University of Malaga, Spain
Dario Ojeda Alayon, Norwegian Institute of Bioeconomy Research, Norway
Copyright © 2022 Feng, Gao, Zhang, Jiang, Li, Deng, Liao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Xu, xubo@cib.ac.cn