 Xue-Ping Wei1,2
Xue-Ping Wei1,2 Xiao-Yi Zhang1
Xiao-Yi Zhang1 Yu-Qing Dong1Ji-Long Cheng1Yun-Jun Bai3
Yu-Qing Dong1Ji-Long Cheng1Yun-Jun Bai3 Jiu-Shi Liu1,2Yao-Dong Qi1,2
Jiu-Shi Liu1,2Yao-Dong Qi1,2 Ben-Gang Zhang1,2
Ben-Gang Zhang1,2 Hai-Tao Liu1,2*
Hai-Tao Liu1,2*- 1Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, China
- 2Engineering Research Center of Tradition Chinese Medicine Resource, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, China
- 3National Resource Center for Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
Most plants of Ligusticum have an important medicinal and economic value with a long history, Ligusticum sinense and L. jeholense (“Gaoben”) has long been used in traditional Chinese medicine for the treatment of carminative, dispelling cold, dehumidification, and analgesia. While in the market Conioselinum vaginatum (Xinjiang Gaoben) is substitution for Gaoben, and occupies a higher market share. These three Gaoben-related medicinal materials are similar in morphology, and are difficult to distinguish from each other by the commonly used DNA barcodes. The chloroplast genome has been widely used for molecular markers, evolutionary biology, and barcoding identification. In this study, the complete chloroplast genome sequences of C. vaginatum, L. sinense, and L. jeholense were reported. The results showed that the complete chloroplast genomes of these three species have typical quadripartite structures, which were comprised of 148,664, 148,539, and 148,497 bp. A total of 114 genes were identified, including 81 protein-coding genes (PCGs), 29 tRNA genes, and four rRNA genes. Our study indicated that highly variable region ycf2-trnL and accD-ycf4 that can be used as specific DNA barcodes to distinguish and identify C. vaginatum, L. sinense, and L. jeholense. In addition, phylogenetic study showed that C. vaginatum nested in Ligusticum and as a sister group of L. sinense and L. jeholense, which suggested these two genera are both in need of revision. This study offer valuable information for future research in the identification of Gaoben-related medicinal materials and will benefit for further phylogenetic study of Apiaceae.
Introduction
Apiaceae is one of the more evolved groups of angiosperms. There are about 250–440 (−455) genera and 3,300–3,700 species in the world, which are widely distributed in temperate regions (Sheh et al., 2005). The highly consistent umbel and two carpels are the most prominent characteristics of Apiaceae, which also makes Apiaceae the first recognized family and easy to identify among angiosperms. However, the high similarity of flower characteristics and dependence on fruit characteristics make it very difficult to identify at genus and species level, and Apiaceae has become a taxonomic difficult group (Constance, 1971).
Plants of Apiaceae play a certain role in the national economy. Many of them can be used as medicinal materials, vegetables, spices, pesticides, and so on. In terms of medicine, such as famous Chinese medicinal materials as Angelica sinensis (Oliv.) Diels, Ligusticum sinense Oliv., Angelica dahurica (Fisch. ex Hoffm.) Benth. et Hook. f. ex Franch. E, Bupleurum chinense DC., etc., these species enjoy a high reputation in the domestic and foreign markets. Ligusticum L. is a widespread, complex genus and the classification of which is controversial. The phylogenetic tree constructed with ITS and chloroplast genes also shows that Ligusticum is polyphyletic, and relationships with nearby genera such as Conioselinum Fisch. ex Hoffm were unclear (Downie et al., 2020).
The traditional Chinese medicine Ligustici Rhizoma et Radix (“Gaoben” in Chinese) is the dried rhizome and root of Ligusticum sinense Oliv. (“Gaoben” in Chinese) and Ligusticum jeholense Nakai et Kitag (“Liao-Gaoben” in Chinese), recorded in the Chinese Pharmacopoeia (Chinese Pharmacopeia Commission, 2020; Figures 1A,B). Medicinal materials Gaoben (in the following, Gaoben represents Ligustici Rhizoma et Radix) has good traditional efficacy for dispelling wind, removing dampness, and relieving pain with a long history (Chinese Pharmacopeia Commission, 2020). Modern pharmacological studies have shown that Gaoben has potential neuroprotective and vasodilation activity and inhibits vascular smooth muscle cell proliferation and anti-inflammatory, analgesic, and antithrombotic activity (Zhang et al., 2001; Lu et al., 2006; Chan et al., 2007; Peng et al., 2007; Lin et al., 2011). Wild resources of Gaoben are gradually decreasing and its yield decreases sharply due to years of excessive collection and destruction of the ecological environment. According to a survey of the medicinal material market, Gaoben occupies a lower share, while Xinjiang Gaoben occupies nearly 70% of the market and is usually used as Gaoben (Zhao, 2007; Figure 1C). In addition, a small amount of other Ligusticum plant was traded as Gaoben (Figure 1D). Xinjiang Gaoben is the dried rhizome and root of the Conioselinum vaginatum (Spreng.) Thell., and is similar in appearance to Gaoben. Such substitutes entered the local medicinal materials market in the 1960 and 1970s and are commonly used as folk medicines in dispelling cold, carminative, and analgesia (Xinjiang Institute of biological soil, Chinese Academy of Sciences, 1981; Pan and Watson, 2005). Source plants of these three Gaoben-related medicinal materials are distributed in different places in China. Wild L. sinense are mainly distributed in Hubei, Sichuan, Shaanxi, Henan, Hunan, Jiangxi, and Zhejiang provinces, and is cultivated in many other provinces and regions. L. jeholense is found mainly in Hebei, Jilin, Liaoning, Shandong, and Shanxi provinces (Chen et al., 1985). Conioselinum vaginatum are grown mainly in mountain meadows, hillside grasses, and river valley shrubs and are distributed in the Tianshan Mountains, Altai Mountains, and western Junggar Mountains in Xinjiang. This species is also distributed in Central Asia, Southwest Asia, Western Siberia, and Central Europe. The three Gaoben-related medicinal materials are similar in morphology, resulting in the difficulty of distinguishing them from each other in appearance. DNA barcode ITS2, which was used to identify Gaoben medicinal materials and their adulterants, can distinguish adulterants from Gaoben, however, it was difficult to distinguish and identify its source plants L. sinense from L. jeholense (Gao et al., 2011).

Figure 1. Gaoben-related medicinal materials in the market (A) Ligusticum sinense, (B) Ligusticum jeholense, (C) Conioselinum vaginatum, and (D) Ligusticum sp.
Chloroplast (cp), an important organelle for photosynthesis in plants, is widely found in terrestrial plants, algae, and some protozoa. It is an important semiautonomous organelle in plant cells with its own genetic material and genetic system (Wolfe et al., 1987). In plants, the chloroplast genome has a conserved quadripartite structure comprising a large single-copy region (LSC) and a small single-copy (SSC) region, which are separated by a pair of inverted repeats (IRs). Most angiosperms have chloroplast genomes ranging in length from 120 to 160 kb (Chumley et al., 2006). Compared with mitochondrial and nuclear genes, chloroplast genomes are often used to explore the origin and evolution of plants, as well as plant genomics and bioinformatics due to their self-replication mechanism and relatively independent evolution (Daniell et al., 2016). The highly conserved structure with a moderate mutation rate of the chloroplast genome makes the comparative analysis suitable for phylogenetic studies at different taxonomic categories, including closely related species (Raubeson and Jansen, 2005; Yi and Choi, 2016).
Because of the different sources of Gaoben and Xinjiang Gaoben, the lack of systematic evaluation of alternative use brings great challenges to the safety and efficacy of clinical medication. In this study, we presented the complete chloroplast genomes of L. sinense, L. jeholense, and C. vaginatum. We aimed to (1) examine the variations including simple sequence repeats (SSRs) and other repeats among these three chloroplast genomes, and (2) discover highly divergent regions of the chloroplast genomes among the source plants of the medicinal materials Gaoben and Xinjiang Gaoben. This study provides beneficial information on the identification of Gaoben-related medicinal materials as well as further phylogenetic studies of Apiaceae.
Materials and Methods
Plant Material, DNA Extraction, and Sequencing
Fresh leaves of L. sinense, L. jeholense, and C. vaginatum were collected from Yuanbao village of Hubei, Qingyuan County of Liaoning, and Nilka County of Xijiang, respectively. All samples were identified by Professor Bengang Zhang, and Dr. Yaodong Qi at the Institute of Medicinal Plant Development (IMPLAD), the Chinese Academy of Medical Sciences (CAMS). The voucher specimens (GB2018052001, LGB2018052501, and XJGB2018080501) were deposited in the Research Center for Medicinal Plant Resources of IMPLAD. Total genomic DNA was extracted from the clean leaves frozen at −80°C using the Plant Genomic DNA Kit following the standard protocol (Tiangen Biotech Co., Ltd., Beijing). DNA quality was assessed based on spectrophotometry and electrophoresis in a 1% (w/v) agarose gel and a NanoDrop spectrophotometer 2000. High-quality DNA was used to generate shotgun libraries with an average insert size of 500 bp and sequenced using Illumina HiSeq X (v2, Illumina, San Diego CA, United States) in accordance with the standard protocol. Approximately, 10 GB of raw data was generated. After the fragments were filtered and trimmed by the fastp program, approximately 4 GB clean reads were obtained for each sample.
Chloroplast Genome Assembly and Annotation
The high-quality paired-end reads were assembled by BLAST++, SSPACE_ Basic_ v2.0.pl. and SOAPdenovo-127mer (Wolfe et al., 1987; Camacho et al., 2009; Boetzer et al., 2011; Luo et al., 2012), and the cp genome of Angelica gigas (accession No.: KT963038) was chosen as the reference. Orientation and circularization of contigs using Geneious (Kearse et al., 2012). Finally, BWA was used to map clean reads to draft the chloroplast genome to ensure that each base was correct (Li and Durbin, 2009). Annotation was performed using the online tools CPGAVAS2 and GeSeq (Tillich et al., 2017; Shi et al., 2019). The genome maps were generated with the OGDRAW program (Lohse et al., 2013). The complete chloroplast genomes were deposited in GenBank with the accession numbers: Ligusticum sinense OM728579, Ligusticum jeholense OM728580, and Conioselinum vaginatum OM728581.
Genome Structure Analysis and Genome Comparison
GC content was calculated using Geneious. The distribution of codon usage was investigated using CodonW with the RSCU ratio. SSRs were determined by using MISA. REPuter was used to identify and locate repeat sequences, including direct, reverse, palindromic, and complement repeats in the chloroplast genomes of three species. The minimal size was 30 bp and the two repeat copies had at least 90% similarity for all repeat types. MAFFT was used to compare the similarity of plastid genome sequences of the six species from Apiaceae (Katoh and Standley, 2013), and mVISTA was used to export visual results to evaluate similarity (Frazer et al., 2004). We also conducted a sliding window analysis to identify the nucleotide variability (Pi) of three Gaoben-related medicinal materials using DnaSP v5.10 (Librado and Rozas, 2009). The step size was set to 200 bp with an 800 bp window length (Zhou et al., 2018).
Species Authentication Assessment
In order to evaluate the species authentication power of the two suggested highly variable regions according to sliding window analysis, we designed specific primers for each region. At the same time we also used two common barcode regions psbA-trnH and ITS2 for comparison (Supplementary Table S1). We collected five batches of medicinal slices from these three species, as well as one unknown species of Ligusticum from the medicine market respectively. We randomly selected three batches from three species and one batch of the unknown species for this study. Eight leaf samples from three species were collected from the field. All of the voucher medicinal materials and voucher specimens were deposited in the Research Center for Medicinal Plant Resources of IMPLAD (Supplementary Table S2). Total genomic DNA was extracted using the Plant Genomic DNA Kit following the standard protocol (Tiangen Biotech Co., Ltd., Beijing). PCR amplification of targeted DNA regions was performed using 12.5 μl 2 × EasyTaq PCR MasterMix (Biomed, Beijing, China), 1 μl each primer (5 μM), and 1–2 μl template DNA and finally replenished with distilled deionized water to the final volume of 25 μl. The purified PCR products were directly sequenced in both directions by Beijing Meijisinuo Biotec. Co., China. Sequences were assembled and aligned using Clustal X v.1.83 (Tompson et al., 1997) and BioEdit v.7.1.11 (Hall, 1999).
Phylogenetic Analysis
In this study, Panax notoginseng was chosen as the outgroup. Chloroplast genomes of 31 species from Apiaceae were downloaded from GenBank (Supplementary Table S3). A total of 58 common protein-coding genes were extracted from 37 chloroplast genomes and the multiple sequences were then aligned using MAFFT v.7 (Katoh and Standley, 2013) and manually adjusted by MEGA 7.0 (Kumar et al., 2018). Phylogenetic analysis was conducted by maximum parsimony (MP), the maximum likelihood (ML), and the Bayesian inference (BI) respectively. MP analysis was carried out in PAUP * 4.0a with heuristic searches of 1,000 random stepwise sequence addition replicates and tree bisection-reconnection (TBR) branch swapping. Bootstrap values (MP-BS) were calculated with 1,000 pseudoreplicates of TBR branch swapping. Before ML and BI analysis, MrModeltest 2.4 was used to determine the best model. ML trees were reconstructed by performing a rapid bootstrap analysis on the RAxML web-server (Stamatakis, 2014), and the optimal nucleotide substitution model was GTR + I + G. BI analysis was carried out in the software package of MrBayes v3.2.1. Four Markov chains were run for 2,000,000 generations and were sampled every 1,000 generations with a random starting tree. After 25% ageing samples were discarded, a consistent tree was built according to the remaining samples, and a posteriori probability was calculated.
Results and Discussion
Features of the Chloroplast Genomes of Gaoben-Related Medicinal Materials
The complete cp genomes of Gaoben-related medicinal materials obtained in this study exhibit a typical circular form with quadripartite structures (LSC, SSC, IRa, and IRb), whose total length was between 148.5–148.7 kb (Figure 2; Table 1). The longest one was C. vaginatum, 148,664 bp. The lengths of L. sinense and L. jeholense were 148,539 and 148,497 bp, respectively. The length of the IR region was between 18.5 and 18.6 kb. Among these species, C. vaginatum, with the longest cp genome, also had the longest IR region, and L. jeholense with the shortest cp genome, and had the shortest IR region. The length of the IR region was consistent with the total length of the chloroplast genome. The reverse repetition of IRa and IRb in the chloroplast genome makes the contraction and expansion of the IR region of most chloroplast genomes positively correlated with the total length.
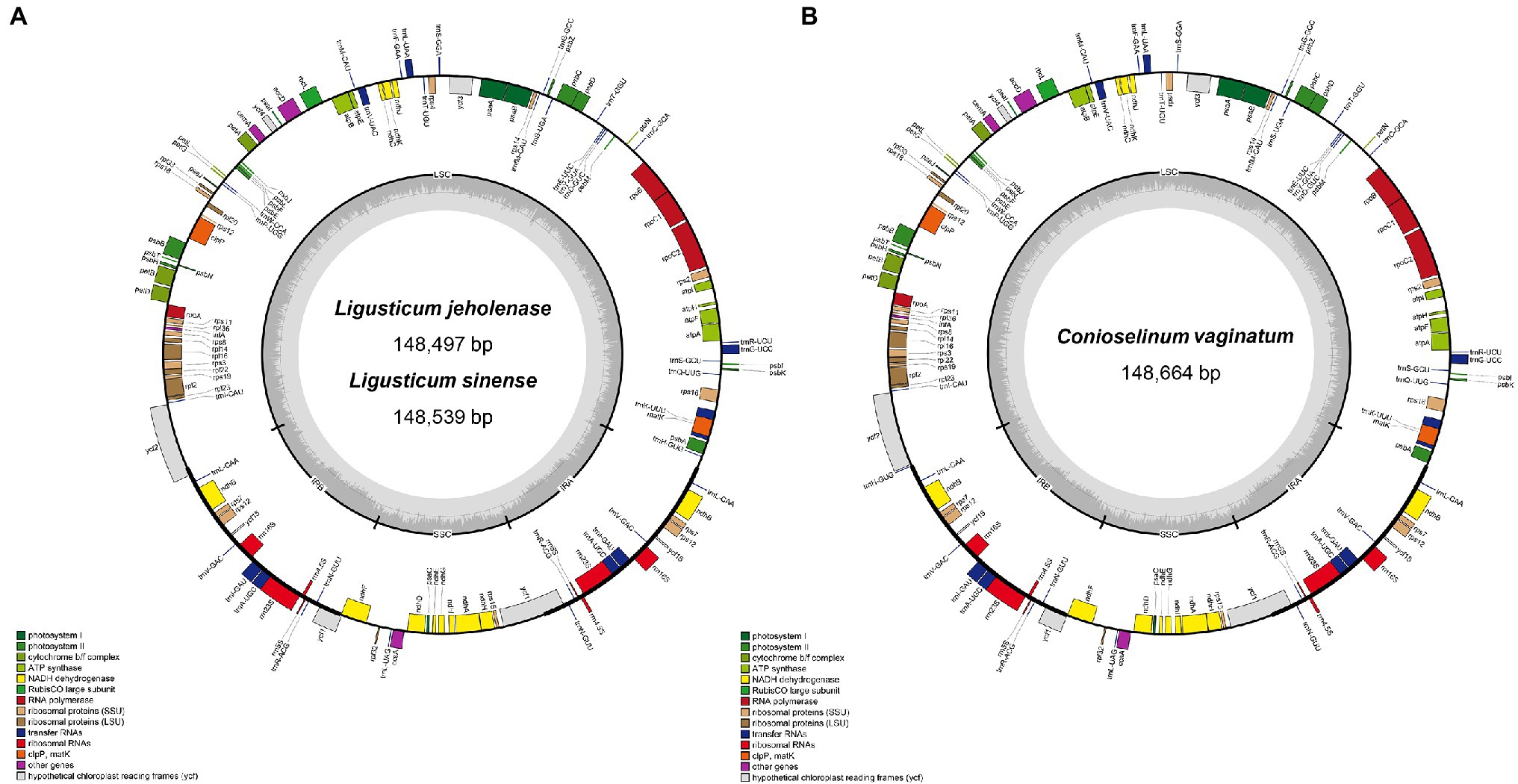
Figure 2. Chloroplast genome maps of (A) Ligusticum sinense and Ligusticum jeholense. (B) Conioselinum vaginatum. Genes drawn inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise. Dark grey shading in the inner circle represents the GC content.
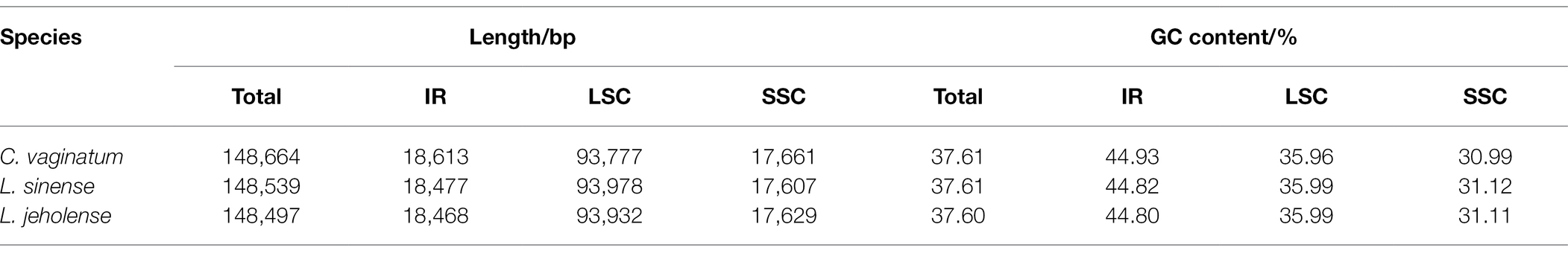
Table 1. Base composition in the chloroplast genomes of Conioselinum vaginatum, Ligusticum sinense, and Ligusticum jeholense.
The GC content of both C. vaginatum and L. sinense was 37.61%, and that of L. jeholense was 37.6%. The GC content of the IR region of these three species was highest, with a range of 44.80 and 44.93%, and the SSC region was lowest, between with a range of 30.99 and 31.12% (Table 1). The high GC content in the IR detected here is similar to most previously reported cp genomes of the angiosperms (Asaf et al., 2020; Wei et al., 2020). The chloroplast genomes of these three species shared the same number and type of genes, and a total of 114 genes were identified, including 81 protein-coding genes (PCGs), 29 tRNA genes, and four rRNA genes. Of these, 15 genes were duplicated in the IR regions (Table 2). A total of 10 genes (rps12, rps16, atpF, rpoC1, petB, petD, rpl16, rpl2, ndhB, and ndhA) and six tRNA genes (trnK-UUU, trnG-UCC, trnL-UAA, trnV-UAC, trnI-GAU, and trnA-UGC) contained one intron, while two genes (ycf3, clpP) contained two introns.
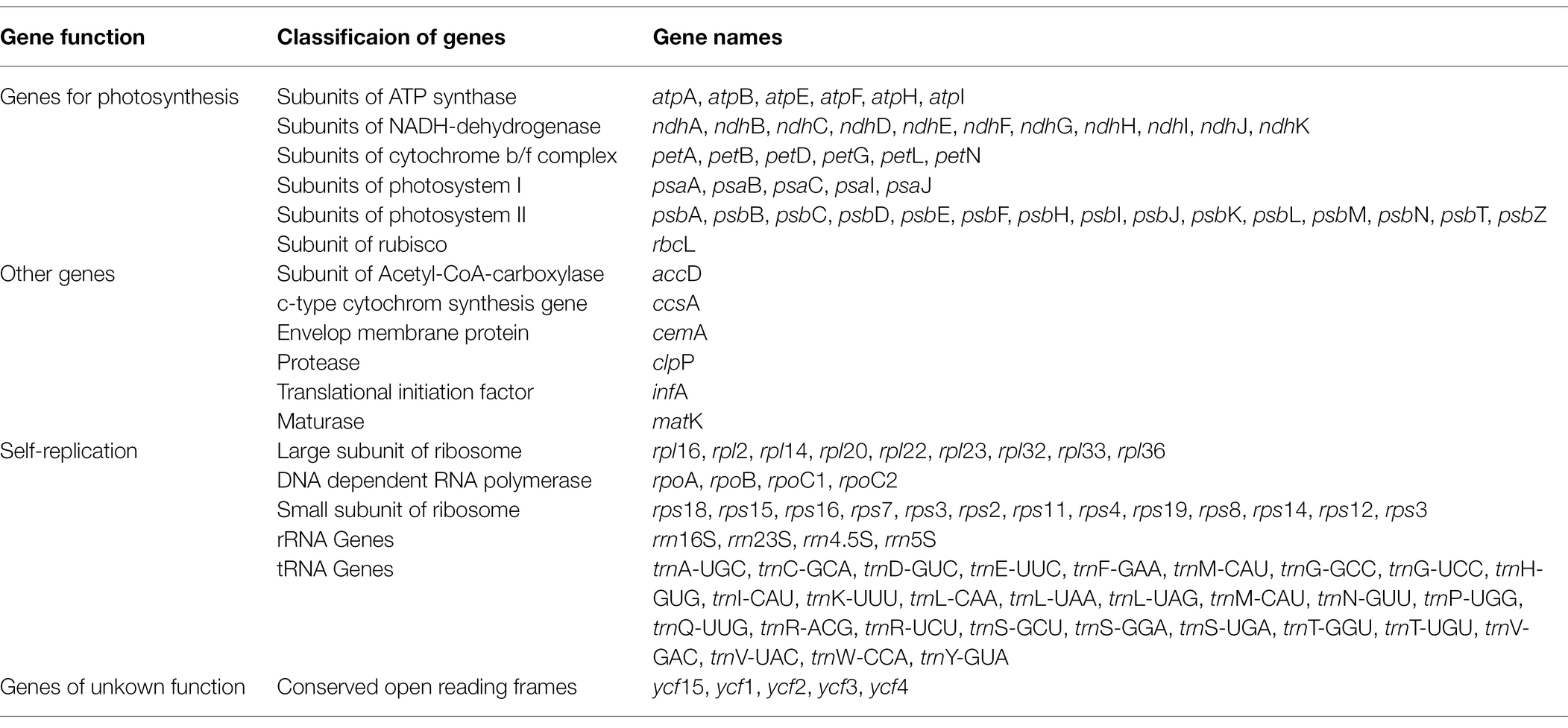
Table 2. Gene contents in the chloroplast genomes of C. vaginatum, L. sinense, and L. jeholense.
Codon Usage Analysis
Codon usage in genomes tends to favor particular subsets of codons. Codon usage bias affects the translational speed and accuracy, and it is associated with the tRNA levels and the GC content in the genomes (Schmid and Flegel, 2011). Relative synonymous codon usage (RSCU) is a measure of nonuniform synonymous codon usage in coding sequences (Wang et al., 2018). Based on the sequences of protein-coding genes (CDSs), the codon usage frequency was estimated for the cp genomes of the three source plants of Gaoben-related medicinal material (Figure 3; Supplementary Table S4). The results revealed the presence of 64 codons, which encode 20 amino acids within the chloroplast protein-coding genes of these three species. All the protein-coding genes were composed of 24,320, 24,392, and 24,343 codons in the cp genomes of C. vaginatum, L. sinense, and L. jeholense, respectively. Leucine and cysteine were the most and least abundant universal amino acids in the cp genomes of the three species, respectively. Except methionine, amino acid codons in the chloroplast genomes of two species preferentially end with A or U (RSCU > 1). The third codon position was biased to end in A and U, accounting for 70.3% of all CDS codons, which is similar to most angiosperm cp genomes (Liu and Xue, 2005). RSCU value analysis showed that most of the amino acid codons have preferences, except methionine and tryptophan, whose codons exhibited no bias (RSCU = 1) in these three cp genomes. Seven codons were over-represented (RSCU > 1.6) in C. vaginatum, while five codons were over-represented in L. sinense and L. jeholense. Twenty codons were underrepresented (RSCU < 1.6) in all three species. High codon preference, especially a strong AT bias in codon usage, is much more common in other plant chloroplast genomes (Wu et al., 2010; Wang et al., 2016).
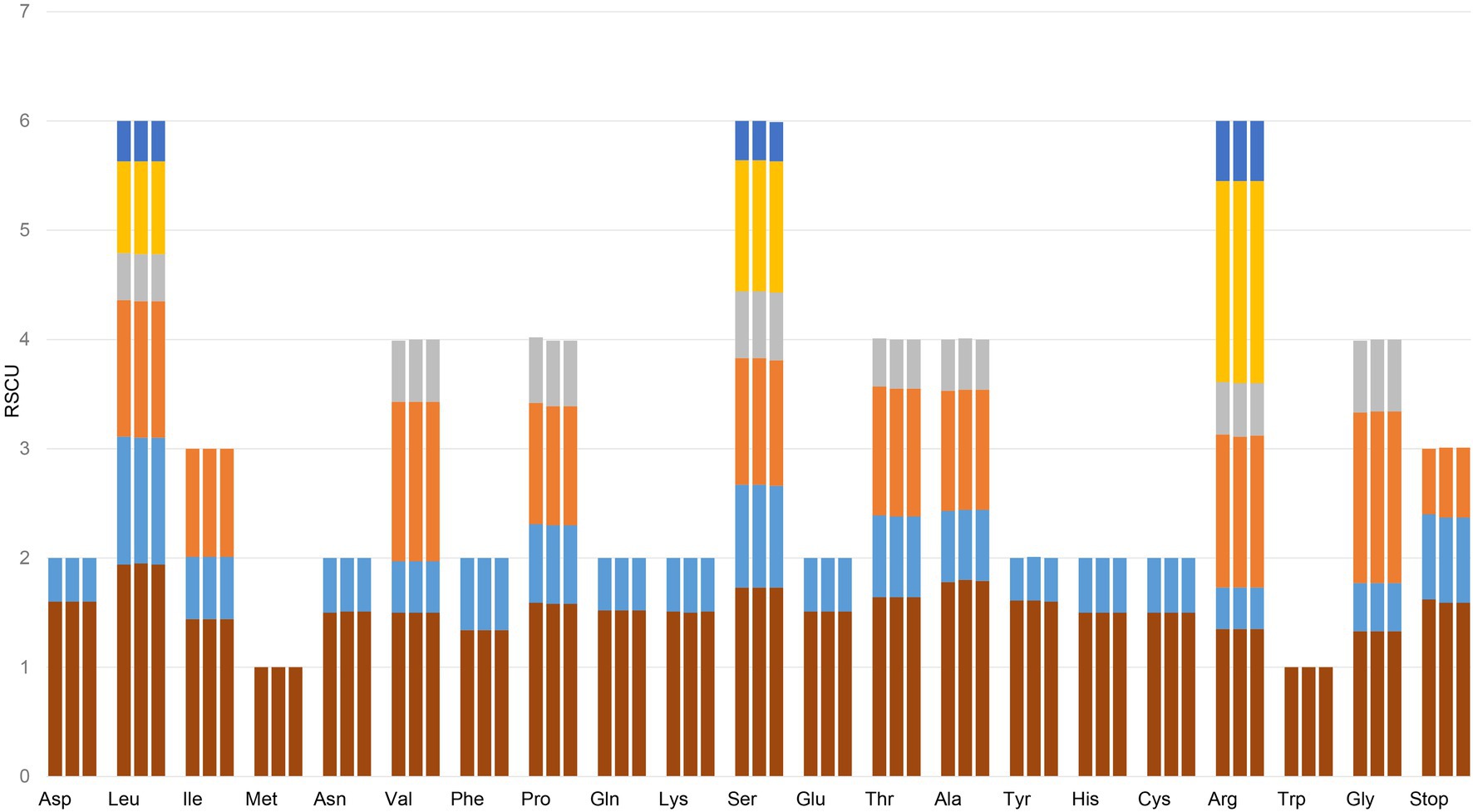
Figure 3. Codon content of 20 amino acid and stop codons in all protein-coding genes of the chloroplast genomes of three species. The histogram from left to right of each amino acid shows codon usage within the chloroplast genomes of C. vaginatum, L. sinense, and L. jeholense.
Simple Sequence Repeats and Repeat Structure Analysis
Simple sequence repeats, also known as microsatellites, are abundantly distributed in the genome. SSRs comprise tandem repeated DNA sequences that consist of 1–6 repeat nucleotide units, and display a high level of polymorphism, placing them as the most preferred genetic markers for species identification, phylogenetic investigations, and other genetic studies (Powell et al., 1995; Yang et al., 2011; Jiao et al., 2012). A total of 84, 78, and 72 SSRs were detected in the cp genomes of C. vaginatum, L. sinense, and L. jeholense, respectively (Table 3), and the number of mononucleotides was between 40 (L. jeholense) and 47 (C. vaginatum). The chloroplast genome of Gaoben-related medicinal material contains 78 SSRs on average, including 44 mono-, 21 di-, 3 tri-, 7 tetra-, 2 penta-, and 1 hexa-nucleotide SSRs. The A/T mononucleotide repeats were the most common. TTA and ATTAG repeats in C. vaginatum were species-specific; AATA, AGATAT, and ATACTG existed in L. sinense and L. jeholense, while AATCA existed in L. sinense and C. vaginatum.
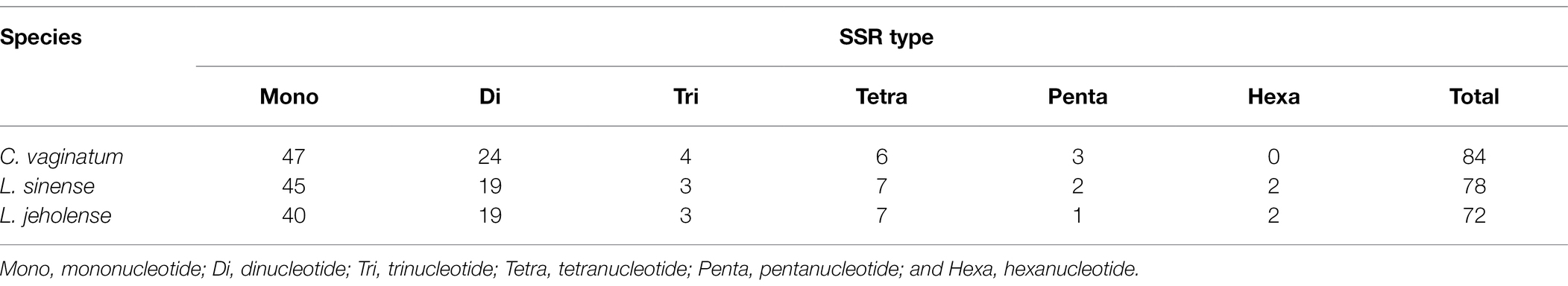
Table 3. Types and amounts of simple sequence repeats (SSRs) in the chloroplast genomes of C. vaginatum, L. sinense, and L. jeholense.
Repeat structures are correlated with rearrangement and recombination of plastomes, which have been used in phylogenetic studies (Inkyu et al., 2017). Repeat sequences with a repeat unit longer than 30 bp were analyzed (Figure 4). The results revealed that the repeats of the chloroplast genome of C. vaginatum had the highest number of dispersed repeats, comprising 25 forwards, 21 palindromic, four reverse repeats, and one complement repeat. L. jeholense had the least dispersed repeats, which contained 18 forwards, 16 palindromic, and one reverse repeat. Most dispersed repeats were distributed in the intergenic regions. The majority of these repeats were mainly forwards and palindromic types with lengths mainly in the range of 30–50 bp. The repeat motif AATCT/AGATT in C. vaginatum was species-specific, ACTGAT/AGTATC and AGATAT/ATATCT existed in L. sinense and L. jeholense, while AAATC/ATTTG existed in L. sinense and C. vaginatum.

Figure 4. Repeat sequences in three chloroplast genomes. F, P, R, and C indicate the repeat types F (forward), P (palindrome), R (reverse), and C (complement), respectively. Repeats with different lengths are indicated in different colors.
Expansion and Contraction of the Border Regions
In the tetrad structure of the chloroplast genome, the IR region is more conserved than the SSC and LSC regions, but in the process of chloroplast evolution, the phenomenon of IR boundary contraction and expansion is widespread in plants (Zhou et al., 2018).
The border regions and adjacent genes of the cp genomes of the three Gaoben-related species were compared to analyze the expansion and contraction variation in junction regions. We selected three closely related species (Carum carvi, Angelica gigas, and Foeniculum vulgare) as references to compare the chloroplast genome structure (Figure 5). All of these species have the IRa/SSC boundary within the ycf1 gene and the IRb/SSC border between the ycf1 gene and ndhF or pseudogenes (ψ) ycf1 and (ψ) ndhF. IRb/LSC and IRa/LSC borders were obvious different in these species: the IRb/LSC boundary of C. vaginatum was in the trnH gene and there are 93 bp from ycf2 gene, while which of L. sinense, L. jeholense, and Angelica gigas were in ycf2 gene. The IRb/LSC boundary of Carum carvi and Foeniculum vulgare was in the rps3 and rpl2 genes, respectively.

Figure 5. The borders of LSC, SSC, and IR regions among six chloroplast genomes. The number above the gene features means the distance between the ends of genes and the borders sites.
Comparative Genome Analysis
The whole chloroplast genome sequences of C. vaginatum, L. sinense, and L. jeholense were compared with those of C. carvi, A. gigas, and F. vulgare using the mVISTA program (Figure 6). The chloroplast genomes of the above species are highly similar. The similarity of L. sinense and L. jeholense was higher, which showed obvious consistency in the positions of 6 k–10 kbp, 13 k–18 kbp, 28 k–34 kbp, 92 k–95 kbp, 115 k–117 kbp, and 147 k–149 bp. In general, the variation of the noncoding region was higher than the variation of the coding region and variation of the variation of the IR region was lower than the variation of the LSC and SSC regions.
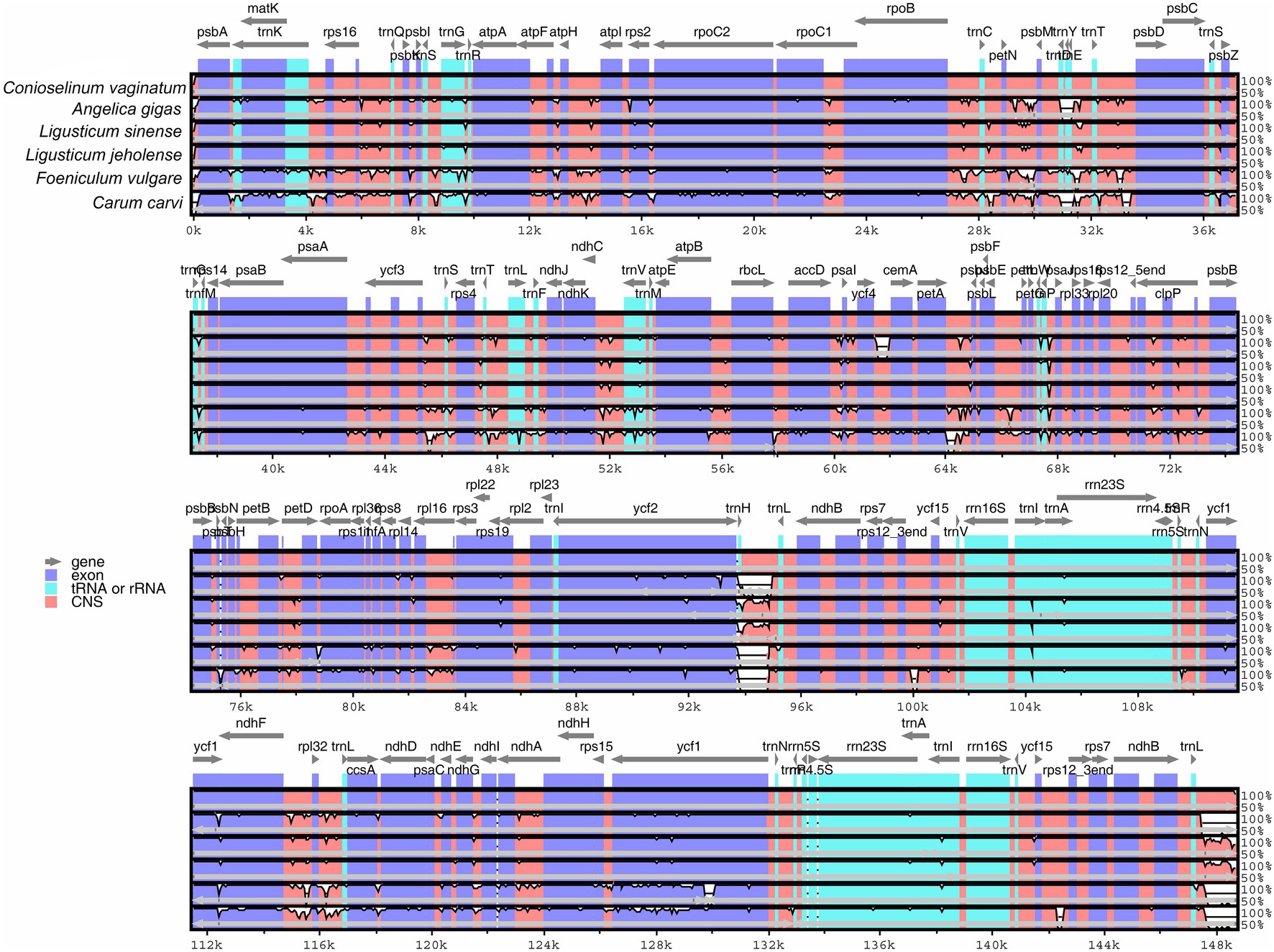
Figure 6. Sequence identity plot comparing the five chloroplast genomes with Conioselinum vaginatum as a reference by using mVISTA. Gray arrows and thick black lines above the alignment indicate genes with their orientation and the position of the inverted repeats (IRs), respectively. A cut-off of 70% identity was used for the plots, and the Y-scale represents the percent identity ranging from 50 to 100%.
Furthermore, sliding window analysis using DnaSP detected highly variable regions in the chloroplast genomes of three Gaoben-related medicinal materials. Nucleotide variability (Pi) was calculated to show divergence at the sequence level (Figure 7). The average value of Pi was 0.002081 among the three Gaoben-related medicinal materials. The IR regions exhibited lower variability than the LSC and SSC regions. A mutational hotspot was observed, which showed remarkably higher Pi values (>0.035) and was located at the IR/LSC boundary. Comparing the nucleotide sequence of mutational hotspot, the variation hotspot was in ycf2-trnL due to trnH gene was inserted between ycf2 gene and trnL gene in C. vaginatum, while trnH gene in L. sinense and L. jeholense was not inserted in ycf2-trnL region, but located between psbA gene and trnL gene. These results suggested that this region might provide the scientific basis for distinguishing C. vaginatum, L. sinense, and L. jeholense (Figure 7A). The average value of Pi was 0.000573 between L. sinense and L. jeholense. The variation hotspot which showed remarkably higher Pi values (>0.007) was located at in LSC regions (Figure 7B). Comparing the nucleotide sequence, the variation hotspot was in the accD-ycf4 region, in which region, there were three SNPs between L. sinense and L. jeholense, and one 7 bp indel (ATAATAA) between C. vaginatum and L. sinense, and L. jeholense (Table 4).
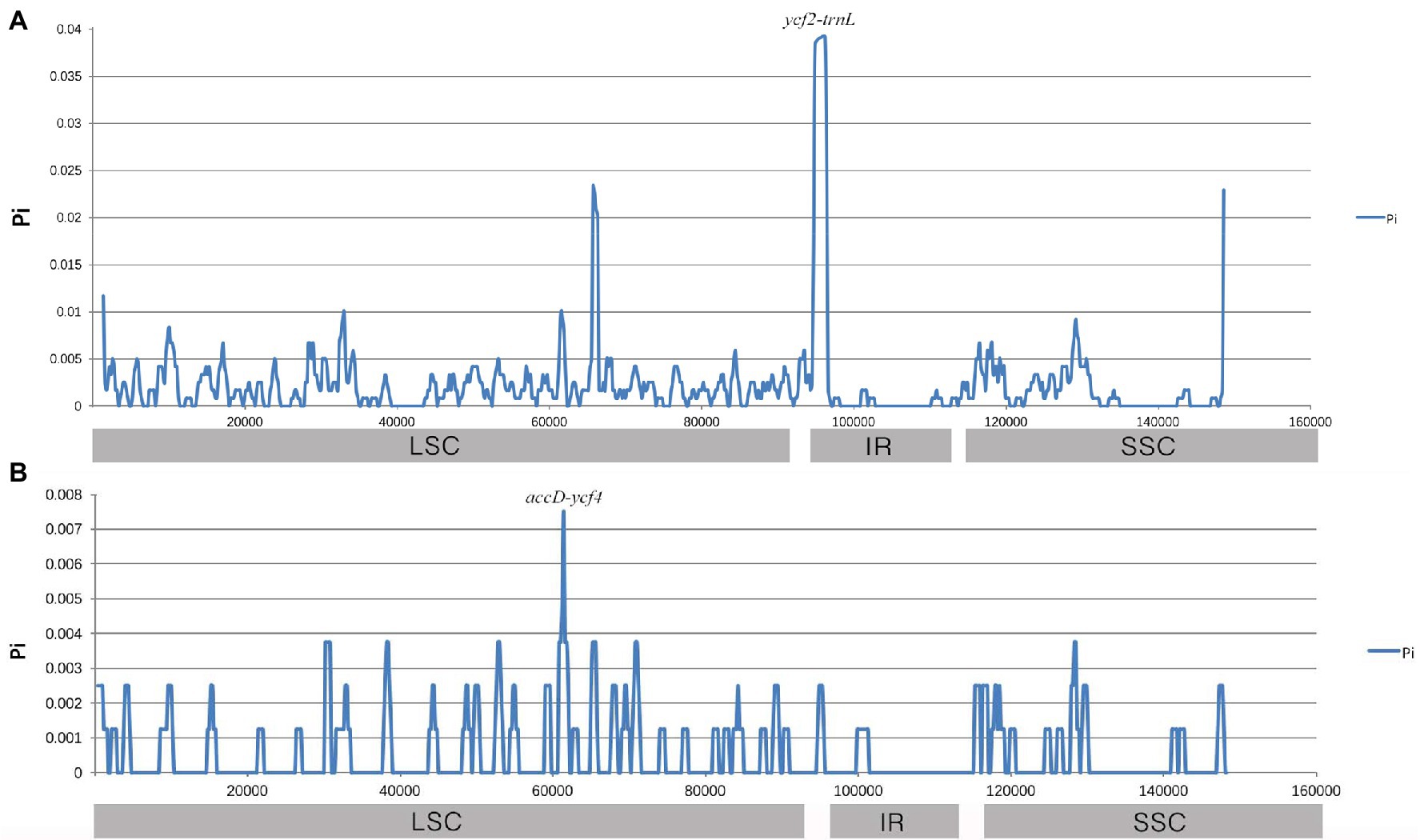
Figure 7. Sliding window analysis of the whole chloroplast genomes. Window length: 800 bp; step size: 200 bp. X-axis: position of the midpoint of a window. Y-axis: nucleotide diversity of each window. (A) Pi among C. vaginatum, L. sinense, and L. jeholense. (B) Pi between L. sinense and L. jeholense.

Table 4. Variation of accD-ycf4 in L. sinense, L. jeholense, and C. vaginatum.
Species Authentication
Two highly variable regions ycf2-trnL and accD-ycf4 as well as two common barcode regions psbA-trnH and ITS2 were amplified and sequenced successfully in both medicinal slices and leaf samples (Supplementary Figure S1). The sequence length of C. vaginatum was significantly longer than that of L. sinense and L. jeholense because of the insertion of trnH gene into ycf2-trnL in C. vaginatum. Sequences of C. vaginatum were 925 bp longer than of L. sinense and L. jeholense. One stability SNP at loci 152 (A/G) of ycf2-trnL can distinguish L. sinense and L. jeholense (Supplementary Data). The 7 bp indel (ATAATAA) at loci 397–403 of accD-ycf4 was stable between C. vaginatum and L. sinense, L. jeholense and can be used to distinguish C. vaginatum. Only one SNP at loci 274 were stable exist between L. sinense and L. jeholense, intraspecific variation was found in the other two SNPs recognized by sliding window analysis (Supplementary Data). The sequences of unknown species from the medicine market were identified as Ligusticum but the species was still uncertain. Previous studies pointed out that the commonly used DNA barcodes ITS2, psbA-trnH and accD were useful in distinguishing L. jeholense from adulterants from other genera but not from the same genus (Gao et al., 2011; Li et al., 2015). In our study, the results showed the similar results that ITS2 and psbA-trnH could separate C. vaginatum from L. sinense and L. jeholense. Ligusticum sinense and L. jeholense had two SNPs in ITS2, but no difference in psbA-trnH (Supplementary Data). Our study suggests that ycf2-trnL and accD-ycf4 could be effective barcodes for distinguishing the three Gaoben-related medicinal materials C. vaginatum, L. sinense, and L. jeholense. Especially, ycf2-trnL can distinguish C. vaginatum from the others based on the results of gel electrophoresis without sequencing. In addition, this study further revealed that the high-variable regions suggested by comparative cp genome analysis can be used as potential molecular markers for species authentication only after verification.
Phylogenetic Analysis
To study the phylogeny of three species of Gaoben-related medicinal material, the protein coding region sequence of the chloroplast genome of 31 species of Apiaceae was analyzed, and Panax notoginseng was selected as the outgroup. The three phylogenetic analyses (MP, ML, and BI) revealed congruent topologies. Three species of Ligusticum and C. vaginatum were recognized as well-supported monophyly (Figure 8, BBMP = 100, BBML = 100, and PPBI = 1). Ligusticum sinense and L. jeholense gathered into a sister group with C. vaginatum, and then with L. tenuissimum, they formed a sister group. The pervious phylogenetic study of Ligusticum inferred from cp genomes and ITS showed that Ligusticum was not monophyly but mixed with others species from Acronema, Sinodielsia, and Selineae (Ren et al., 2020). Our study revealed that Conioselinum embedded Ligusticum which is consistent with the previous phylogenetic studies of the subfamily Apioideae that Ligusticum and Conioselinum were clearly not monophyletic. These two genera are both in need of revision (Downie et al., 2000). Extensive sampling and more robust molecular data are necessary to understand the phylogenetic relationships within Apiaceae (Wen et al., 2020).
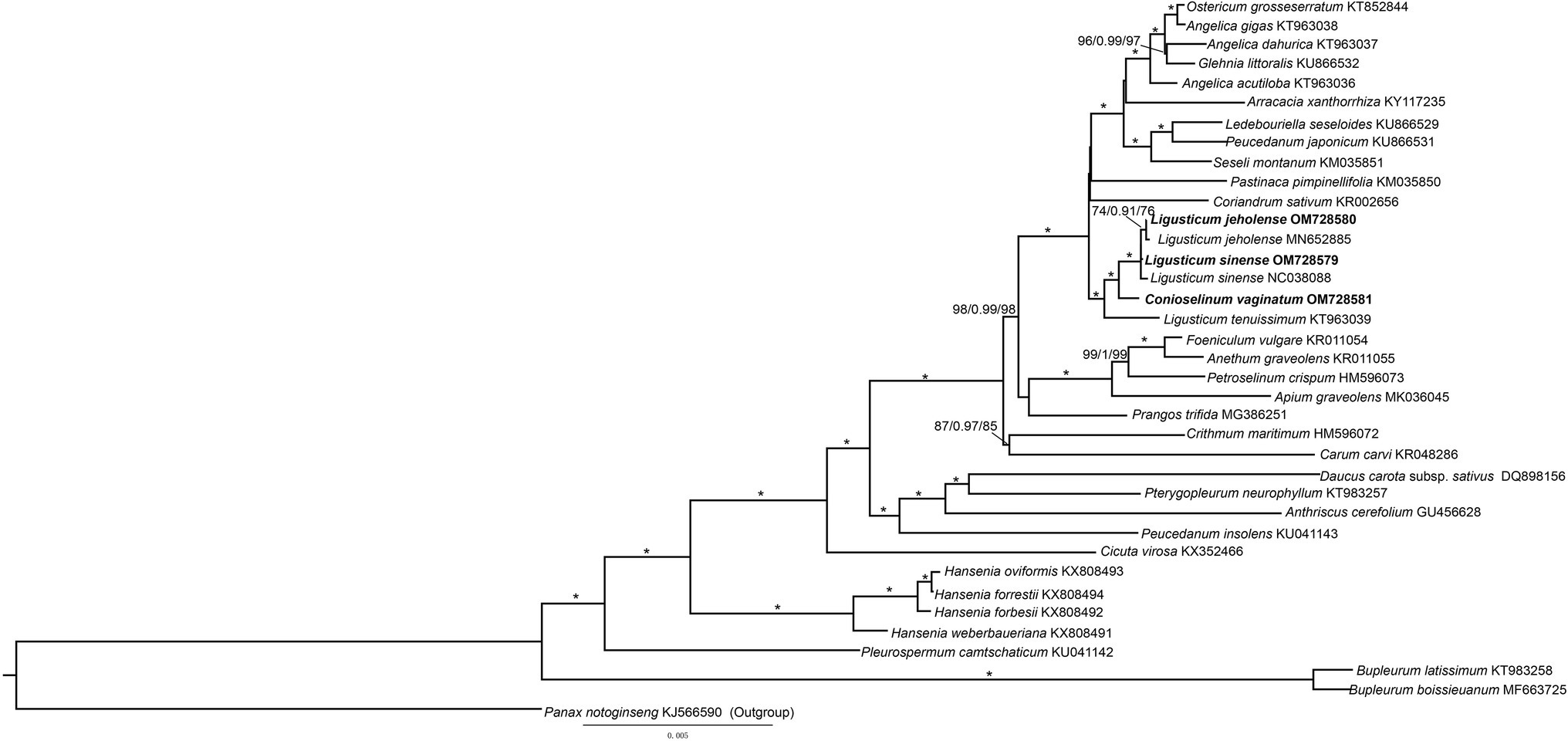
Figure 8. Phylogenetic tree of Apiaceae obtained from the maximum likelihood (ML) analysis of chloroplast genomes. Numbers on branches are support values (BSML/PPBI/BSMP), Stars indicate BSMP, BSML = 100%, and PPBI = 1.0.
Conclusion
The complete chloroplast genomes of three Gaoben-related medicinal materials were determined in this study. The results revealed that genome size, structure, gene content, as well as compositional organization were highly conserved among the three species. Species of Ligusticum and C. vaginatum were recognized as well-supported monophyly, which revealed the closely related relationship between Ligusticum and Conioselinum. Our study suggested that ycf2-trnL and accD-ycf4 could be effective barcodes for distinguishing the three Gaoben-related medicinal materials C. vaginatum, L. sinense, and L. jeholense. This study provided a new method for the accurate identification of three Gaoben-related medicinal materials and provided a scientific basis for differential evaluation of Gaoben-related medicinal materials.
Data Availability Statement
The complete cp genome sequences of Ligusticum sinense, Ligusticum jeholense, and Conioselinum vaginatum are deposited in GenBank, accession number: OM728579, OM728580, and OM728581.
Author Contributions
X-PW was responsible for data analysis and writing of the manuscript. X-YZ and Y-QD performed the bioinformatics work. X-PW, Y-JB, and J-LC did experimental work and data analyses. J-SL participated in the production of figures. Y-DQ and B-GZ collected and identified samples. H-TL conceived the design of the study. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by National Natural Science Foundation of China (Grant No. 81673543) and the State Key R&D Program on “Modernization of Traditional Chinese Medicine” (2018YFC1707904).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Mr. Xinlei Zhao for his kind help in collecting the samples of medicinal materials. We would also thank the two reviewers for their constructive comments and suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.878263/full#supplementary-material
References
Asaf, S., Khan, A. L., Khan, A., and Al-Harrasi, A. (2020). Unraveling the chloroplast genomes of two Prosopis species to identify its genomic information, comparative analyses and phylogenetic relationship. Int. J. Mol. Sci. 21:3280. doi: 10.3390/ijms21093280
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). Blast+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chan, S. K., Cheng, T. Y., and Lin, G. (2007). Relaxation effects of ligustilide and senkyunolide A, two main constituents of Ligusticum chuanxiong, in rat isolated aorta. J. Ethnopharmacol. 111, 677–680. doi: 10.1016/j.jep.2006.12.018
Chen, S. K., Chen, B. Y., Li, H., and Ligusticum, L. (1985). Delectis Florae Reipublicae Popularis Sinicae Agendae Academiae Sinicae Edita, Flora Reipublicae Popularis Sinicae 55. Beijing: Science Press, 252.
Chinese Pharmacopeia Commission (2020). Pharmacopoeia of the People’s Republic of China. Beijing: Chinese Medical Science and Technology Press, 397.
Chumley, T. W., Palmer, J. D., Mower, J. P., Matthew, F. H., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of pelargonium×hortorum: organization and evolution of the largest and most highly rear ranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Constance, L. (1971). History of the classification of Umbelliferae (Apiaceae). Bot. J. Linn. Soc. 64, 1–11.
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Downie, S. R., Katz-Downie, D. S., and Watson, M. (2000). A phylogeny of the flowering plant family Apiaceae based on chloroplast DNA rpl16 and rpoC1 intron sequences: towards a suprageneric classification of subfamily Apioideae. Am. J. Bot. 87, 273–292. doi: 10.2307/2656915
Downie, S. R., Watson, M. F., Spalik, K., and Katz-Downie, D. S. (2020). Molecular systematics of Old World Apioideae (Apiaceae): relationships among some members of tribe Peucedaneae sensu lato, the placement of several island-endemic species, and resolution within the apioid superclade. Can. J. Bot. 78, 506–528. doi: 10.1139/cjb-78-4-506
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Inna, D. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, T., Yao, H., and Chen, S. L. (2011). Molecular identification of Ligustici Rhizoma et radix and ITS adulterants based on ribosomal DNA ITS2 sequence. World Sci. Technol. 13, 418–423. doi: 10.3969/j.issn.1674-3849.2011.02.038
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Inkyu, P., Yang, S., Goya, C., Wook, K., and Byeong, M. (2017). The complete chloroplast genome sequences of aconitum pseudolaeve and aconitum longecassidatum, and development of molecular markers for distinguishing species in the aconitum subgenus Lycoctonum. Molecules 22:2012. doi: 10.3390/molecules22112012
Jiao, Y., Jia, H. M., Li, X. W., Chai, M. L., and Gao, Z. S. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genomics 13:201. doi: 10.1186/1471-2164-13-201
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, X., Qin, Y. T., and Dong, X. H. (2015). DNA barcode identification of Ligusticum jeholense and its adulterants. J. Northeast. Agric. Univ. 46, 22–31. doi: 10.3969/j.issn.1005-9369.2015.05.004
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin, Q., Zhao, A. G., Chen, J. N., Lai, X. P., Gui, S. H., and Fang, C. P. (2011). Anti-inflammatory and analgesic effects of ligustilie. Chin. J. Exper. Tradit. Med. Formul. 17, 165–168. doi: 10.3969/j.issn.1005-9903.2011.11.047
Liu, Q., and Xue, Q. (2005). Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 84, 55–62. doi: 10.1007/BF02715890
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). Organellar genome DRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Lu, Q., Qiu, T. Q., and Hong, Y. (2006). Ligustilide inhibits vascular smooth muscle cells proliferation. Eur. J. Phamacol. 542, 136–140. doi: 10.1016/j.ejphar.2006.04.023
Luo, R. B., Liu, B. H., Xie, Y. L., Li, Z. Y., Huang, W. H., Yuan, J. Y., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18. doi: 10.1186/2047-217X-1-18
Pan, Z. H., and Watson, M. F. (2005). “Conioselinum Fischer ex Hoffmann,” in Flora of China, Beijing, Science Press. eds. Z. Y. Wu and P. H. Peter, Vol. 14 (St. Louis: Missouri Botanical Garden Press), 155–156.
Peng, H. Y., Du, J. R., Zhang, G. Y., Kuang, X., and Liu, Y. X. (2007). Neuroprotective effect of Z-ligustilide against permanent focal ischemic damage in rats. Biol. Pharm. Bull. 30, 309–312. doi: 10.1248/bpb.30.309
Powell, W., Morgante, M., Mcdevitt, R., Vendramin, G. G., and Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U. S. A. 92, 7759–7763. doi: 10.1073/pnas.92.17.7759
Raubeson, L. A., and Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Chloroplast Genomes of Plants, Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants. ed. R. J. Henry (United Kingdom: CABI publishing), 45–68.
Ren, T., Li, Z. X., Xie, D. F., Gui, L. J., Peng, C., Wen, J., et al. (2020). Plastomes of eight Ligusticum species: characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol. 20:519. doi: 10.1186/s12870-020-02696-7
Schmid, P., and Flegel, W. A. (2011). Codon usage in vertebrates is associated with a low risk of acquiring nonsense mutations. J. Transl. Med. 9:87. doi: 10.1186/1479-5876-9-87
Sheh, M. L., Pu, F. D., Pan, Z. H., Watson, M. F., Cannon, J. F. M., Holmes-Smith, I., et al. (2005). “Flora of China,” in Apiaceae. ed. Flora of China Editorial Committee (St. Louis, Missouri, USA: Missouri Botanical Garden Press)
Shi, L. C., Chen, H. M., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Wang, S., Yang, C. P., Zhao, X. Y., Chen, S., and Qu, G. Z. (2018). Complete chloroplast genome sequence of Betula platyphylla: gene organization, RNA editing, and comparative and phylogenetic analyses. BMC Genomics 19:950. doi: 10.1186/s12864-018-5346-x
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016). Complete chloroplast genome sequence of Aquilaria sinensis (lour.) Gilg and evolution analysis within the Malvales order. Front. Plant Sci. 7:280. doi: 10.3389/fpls.2016.00280
Wei, X. P., Li, H. J., Che, P., Guo, H. J., Zhang, B. G., Liu, H. T., et al. (2020). Comparing chloroplast genomes of traditional Chinese herbs Schisandra sphenanthera and S. chinensis. Chin. Herb. Med. 12, 247–256. doi: 10.1016/j.chmed.2019.09.009
Wen, J., Yu, Y., Xie, D. F., Peng, C., Liu, Q., Zhou, S. D., et al. (2020). A transcriptome-based study on the phylogeny and evolution of the taxonomically controversial subfamily Apioideae (Apiaceae). Ann. Bot. 125, 937–953. doi: 10.1093/aob/mcaa011
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U. S. A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Wu, F. H., Chan, M. T., Liao, D. C., Hsu, C. T., Lee, Y. W., Daniell, H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10:68. doi: 10.1186/1471-2229-10-68
Xinjiang Institute of biological soil, Chinese Academy of Sciences (1981). Flora of Medicinal Plants in Xinjiang 102. Urumchi: Xinjiang Publishing.
Yang, A. H., Zhang, J. J., Yao, X. H., and Huang, H. W. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 98, 123–126. doi: 10.3732/ajb.1000532
Yi, D. K., and Choi, K. (2016). The complete chloroplast genome sequence of Abies nephrolepis (Pinaceae: Abietoideae). J. Asia Pac. Biodivers. 9, 245–249. doi: 10.1016/j.japb.2016.03.014
Zhang, M. F., Shen, Y. Q., Zhu, Z. P., and Wang, H. W. (2001). Anti-thrombotic, choleretic and anti-ulcerous actions of Rhizoma Ligustici. Chin. Pharm. 12, 329–330. doi: 10.3969/j.issn.1001-0408.2001.06.004
Zhao, W. (2007). Study on Cultivation Techniques of Ligusticum Jeholense. Jilin: Jilin Agricultural University.
Keywords: Apiaceae, chloroplast genome, Conioselinum vaginatum, DNA barcode, Ligusticum jeholense, Ligusticum sinense, phylogeny
Citation: Wei X-P, Zhang X-Y, Dong Y-Q, Cheng J-L, Bai Y-J, Liu J-S, Qi Y-D, Zhang B-G and Liu H-T (2022) Molecular Structure and Phylogenetic Analyses of the Complete Chloroplast Genomes of Three Medicinal Plants Conioselinum vaginatum, Ligusticum sinense, and Ligusticum jeholense. Front. Plant Sci. 13:878263. doi: 10.3389/fpls.2022.878263
Edited by:
Robert Philipp Wagensommer, University of Bari Aldo Moro, ItalyReviewed by:
Abdullah, Quaid-i-Azam University, PakistanYun-peng Du, Beijing Academy of Agricultural and Forestry Sciences, China
Copyright © 2022 Wei, Zhang, Dong, Cheng, Bai, Liu, Qi, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai-Tao Liu, htliu0718@126.com