
Abstract
The new labdane-type diterpenoid, physaglutinic acid (1), was isolated from the hexane extract of Physalis glutinosa. This compound is the first succinyl labdane isolated from a Physalis species. The known constituents of this plant were identified as the labdanes 12-O-acetylphysacoztomatin (3), physacoztomatin (4) and 12-epi-nicandrodiol (6). The flavonol retusin (8) was also isolated from this species. The investigation of the acetone extract of Physalis latiphysa resulted in the isolation of the known (+)- (Z)-labda-8(17),13-diene-15,16-diol (9), labdenediol (11), the epimeric mixture of physanicantriol (12) and 14 epi-physanicantriol (13), together with the sucrose esters, nicandroses B (15) and D (16). Also, a large amount of the flavonol glycoside rutin (17) was obtained from the methanol extract. The activity of eight of the isolated compounds and three of their derivatives as lipase inhibitors was determined. The mode of binding of active compounds 4, 10 and 16 was explored using molecular docking on the binding pocket of pancreatic lipase (PDB ID 1LPB).

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A revision of the literature regarding the constituents of Physalis species (family Solanaceae) reveals that the most frequently isolated metabolites from these plants are ergostane-type steroids with a δ or γ-lactone in the side chain, which are known as withanolides [1, 2]. However, in some species of this genus, such as P. coztomatl [3], P. sordida [4], and P. nicandroides var. attenuatta [5], labdane-type diterpenoids, as compounds 1 to 14, are some of their major constituents. Even more, despite a detailed study of P. nicandroides [6], withanolides were not detected and only a series of labdanes and sucrose esters were isolated. Now, as continuation of our investigation of Physalis species, we carried out the studies of P. glutinosa Schltdl. and P. latiphysa Waterf. that led to the isolation of labdanes, sucrose esters, and flavonoids, and in which the presence of withanolides was not detected either. It should be noted that among the compounds isolated from P. glutinosa we found the first succinyl-labdane reported from a Physalis species. This is interesting since succinyl-labdanes are present in the Araucariaceae, Leguminosae and Asteraceae families [7, 8], but also in the polylabdanoid Class Ia amber or resinites [9], which were formed by polymerization of plants resins throughout millions of years, and in which the role of succinic acid is to cross-link their macromolecular structure [10].
On other hand, obesity is one of the major threats to worldwide health; it is estimated that more than one billion people in the planet are obese and this state is associated with several metabolic disorders such as diabetes mellitus, cardiovascular diseases, dyslipidemia, and cancer, among others [11]. One of the therapeutical approaches to obesity consists of inhibiting pancreatic lipase, which hydrolyses triglycerides into fatty acids and glycerol, thus allowing intestinal cells to absorb dietary fats. Since orlistat is the only lipase inhibitor approved by the Food and Drug Administration (FDA), there is a need for novel drugs targeting this enzyme [12, 13]. As far as we know, P. glutinosa and P. latiphysa do not have a documented history of medicinal uses, but considering that several labdanes and their derivatives have shown potential to inhibit pancreatic lipase [14, 15], it was decided to evaluate the in vitro lipase inhibitory activity of six of the isolated labdanes, as well as of three of their derivatives. To explore other possible types of drug candidates, a flavonoid and a sucrose ester were included in the bioassay (Fig. 1).

A Structures of the natural compounds from P. glutinosa (1, 3, 4, 6 and 8) and their derivatives 2 and 7. B Structures of the natural compounds from P. latiphysa (9, 11–13 and 15–17) and their derivatives 10 and 14
Results and discussion
Compound 1, isolated as a colorless viscous liquid, exhibited a broad IR band between 2800 and 3600 cm−1, indicative of a carboxylic acid, and another band at 1731 cm−1 that was attributed to the carbonyls of the acid and/or a saturated ester. Its molecular formula was determined as C26H40O6 by the [M + NH4]+ ion peak at m/z 466.31965, and by the 26 carbon signals in the 13C NMR spectrum (Table 1). Two of these signals (δC 170.8 and 21.3) correspond to an acetyl group (δH 2.09), while those of two methylene carbons (δC 29.0 and 28.8; δH 2.65 m, 4H), an acid (δC 176.1), and an ester (δC 172.0) carbonyl signals, indicated the presence of a succinyl group. The presence of the free carboxylic acid was probed by the obtention of the methyl ester (2) whose NMR spectra showed the signals for the OMe group (δC 51.8; δH 3.69) and the high field shift of the new ester carbonyl (δC 172.7, C-4”). The remaining NMR signals of compound 1 were attributed to a labda-7,13(E)-diene with oxygenated functions at C-12 and C-15. Thus, the presence of the double bond at C-7 was deduced from the signals for CH-7 (δC 123.0; δH 5.41), C-8 (δC 134.3), and CH3-17 (δC 22.3; δH 1.72), while that at C-13 was consistent with the signals for C-13 (δC 140.4), CH-14 (δC 125.1; δH 5.57), CH2O-15 (δC 60.9; δH 4.65), and CH3-16 (δC 22.3; δH 1.72). The HMBC spectrum showed the correlations of H-14 with the carbons of the oxymethine at C-12 (δC 78.4; δH 5.28) and of CH3-16. The HMBC correlation of H-12 with the carbonyl carbon of the acetyl group and that of H2-15 with C-1’of the succinyl group determined the position of these groups. The E-configuration of the C-13-double bond was based on the NOE effects between H2-15 and H3-16 in the NOESY spectrum (Fig. 2). Previously, it was observed that in 12-hydroxylabdanes with a vinyl C-8, the C-12-configuration affects the chemical shift of the H-9 signal, which in (12 R)-hydroxy compounds resonates at ∼δH 2.05 and at ∼δH 1.70 in their 12-O-acetyl derivatives, while in the (12 S)-hydroxy compounds, it appeared at ∼δH 1.50 and at ∼δH 1.40 in their 12-O-acetyl derivatives [5]. In compound 1, this signal was observed at δH 1.72, thus suggesting a (12 R)-configuration. Supporting this assumption, a close similarity was observed between the NMR spectra of compounds 1 and 12,15-di-O-diacetylphysacoztomatin (5) [4]. In fact, except for the signals of the different C-15-O-acyl groups, the NMR spectra of 1 and 5 were practically superimposable. Compound 5 was isolated from P. sordida and it was also obtained upon acetylation of physacoztomatin (4), whose structure was confirmed by X-ray analysis [3, 4]. In this way the structure of 1, named physaglutinic acid, was established as (12 R)-O-acetyl-15-O-succinyl-labda-7,13(E)-diene.

Key HMBC and NOESY correlations for compound 1
Besides of 1, the known labdanes 12-O-acetylphysacoztomatin (3), physacoztomatin (4) [3, 4] and 12-epi-nicandrodiol (6) [5] together with the flavonol retusin (8) [16] were isolated from P. glutinosa (Fig. 1A). It is important to highlight the unusual high concentration of compound 6 in the plant (1.49 % with respect to the weight of dry plant material).
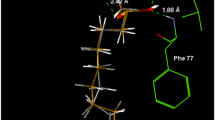
Regarding to the study of Physalis latiphysa Waterf., the examination of the acetone extract led to the isolation of the known compounds (+)-(Z)-labda-8(17)-13-diene-15,16-diol (9) [17], labdenediol (11) [18, 19], a mixture of the epimers physanicantriol (14 S) (12) and 14 epi-physanicantriol (14 R) (13) [6], and nicandroses B (15) and D (16) [20], while the flavonol glycoside rutin (17) [21] was obtained from the methanol extract (Fig. 1B). The chemical profile found in this species was very similar to the one found in P. nicandroides [6]. The structural elucidation of all the known compounds isolated from both Physalis species was carried out by analysis of their physical and spectroscopic data, which were compared with those reported. In the case of compound 9, its NMR data recorded in CDCl3 were used for comparison, however, since it is sensitive to acid, its spectra were acquired in acetone-d6 (Table 2). In this solvent, some peculiarities such as duplicated signals for C-15 and C-16, and the integral for the proton signal (t, J = 5.5 Hz) of each of the hydroxyl groups bonded to CH2-15 and CH2-16 that corresponds to a half proton, were observed. An explanation for these facts is the existence of two different hydrogen bonds which are stabilized by acetone (Fig. 3).

Stabilized intramolecular hydrogen bonds of compound 9
Since another objective of this work was to discover new inhibitors of pancreatic lipase and considering that labdanes such as the dialdehyde (E)-labda-8(17),12-diene-15,16-dial have this activity (IC50 14.63 μM) [14], six of the isolated labdanes were evaluated. Also, in order to explore a greater structural diversity, derivatives 7 [22], 10, and 14 [23] were prepared and included in the bioassay, together with the flavonoid 8 and the sucrose ester 16.
The activity was assessed using a spectrophotometric method with some modifications [24, 25], with concentrations ranging from 0.3 to 300 μM. As seen in Table 3, physacoztomatin (4) and diacetyl derivative (10) inhibited the enzyme, although the effects were not concentration dependent. Maximal inhibition was achieved at 50 μM (24.89 ± 4.29 and 19.57 ± 2.9%, respectively). Noteworthy, the sucrose ester, nicandrose D (16), showed a concentration-dependent inhibition when evaluated between 0.3 and 300 μM, thus, it was evaluated using concentrations between 200 and 500 μM. Maximal inhibition achieved was 64.88 ± 0.93% at 500 μM, whilst calculated IC50 was 392.10 ± 0.02 μM. Compounds 6−9 and 11−14 were inactive from 0.3 to 300 μM. For its part, orlistat, used as positive control reached 75.67 ± 1.67% at 100 µM, but its calculated IC50 was 1.22 ± 0.2 μM. Therefore, although having a high efficacy, nicandrose D is a much less potent lipase inhibitor than orlistat.
To gain further insight on the mode of binding of active compounds 4, 10 and 16, a molecular docking analysis was conducted. Figure 4A shows the lipase-colipase complex and the catalytic site, where dockings were simulated. As depicted in Fig. 4B, the active compounds fit in the catalytic site in a similar manner as the co-crystallized inhibitor. As expected, the docked compounds interact with at least two residues (Ser152, His263) of the catalytic triad (Ser152, Asp176, and His263). Some of these interactions are stabilized by one or two hydrogen bonds, conferring a better docking score. In this study, the positive control orlistat ranks as the ligand with the highest docking score (−8.50 kcal/mol), followed by 16 (−8.35 kcal/mol), 10 (−7.16 kcal/mol), and 4 (−7.02 kcal/mol), in agreement with our in vitro data. A possible explanation of these results is that unlike 4 and 10, nicandrose D (16) holds a C10 aliphatic ester which resembles the hydrophobic chain of the alkyl phosphonate (previously co-crystallized with lipase) [26], whose docking score was −6.79 kcal/mol. Thus, increasing the length of the chain seems to increase the binding affinity [27]. This observation also agrees with our experimental results, as compounds 4 and 10 are lacking this moiety and are less effective lipase inhibitors.

Crystal structure of the lipase-colipase complex (PDB ID: 1LPB). A Lipase-colipase complex (cyan ribbons represents the lipase, magenta ribbons represent colipase moiety) with the docked molecules (yellow sticks for query molecules, blue sticks for co-crystallized inhibitor). B Detailed image of the site of interaction, showing the docked molecules into the catalytic pocket, depicted a line-surface representation (green surface indicates hydrophobic interactions, blue surface indicates polar interactions and red surface specifies exposed residues)
Table 4 shows the resulting protein-ligand interactions of the docking simulations, and Fig. 5 depicts the docking poses of analyzed lipase inhibitors. Orlistat acts by binding covalently to the serine residues on the active sites of both gastric and pancreatic lipase, thus preventing them from interacting with its substrate [26]. The amino acids involved in orlistat’s inhibition are Ser152, Asp176, and His263.

Relevant interactions of compounds (cyan sticks, surface) 4 (A), 10 (B), 16 (C), and orlistat (D) with lipase protein (PDB ID 1LPB). In all cases, the interaction pocket is represented as white sticks, highlighting catalytic triad residues (Ser152, Asp176 and His263) as orange sticks. Hydrogen bonds are shown as green dotted lines
Several sucrose esters isolated from other Solanaceae species have demonstrated a variety of biological effects, such as antibacterial and antifungal, and others associated to ecological interactions (antifeedant, antiovipositional, insecticidal, molluscicidal, aphicidal). Noteworthy, the hypoglycemic activity of peruviosides A-F, sucrose esters isolated from Physalis peruviana, has been related to their ability to inhibit intestinal α-amylase, an enzyme responsible for the hydrolysis of carbohydrates [27, 28]. Furthermore, structure-activity relationships showed that the length of the C-2 fatty acid of sucrose affects the inhibitory activity. This relationship for lipase inhibitory effect needs to be evaluated for other sucrose esters, but our results suggest the potential of these natural products to act as a novel class of lipase inhibitors.
Conclusions
Physaglutinic acid (1) was isolated from Physalis glutinosa. This new compound is the first succinyl labdane isolated from a Physalis species. Labdane diterpenoids were the main constituents of P. glutinosa and P. latiphysa; flavonoids were also present in both species, while sucrose esters only were found in P. latiphysa. Withanolides were not detected in either of the two species, increasing to three the number of Physalis species that produce labdanes, but not withanolides. In bioassays that evaluated the ability of nine labdanes, one flavonoid, and one sucrose ester to inhibit pancreatic lipase activity, only labdanes 4 and 10 and sucrose ester 16 were active. The highest activity corresponded to 16. Docking analysis seems to support the hypothesis that the length of the ester chain in C-2 of glucose is important for the lipase inhibitory activity.
Experimental
General experimental procedures
Melting points were determined on a Fischer-Johns melting point apparatus. Optical rotations were measured on a Perkin Elmer 343 polarimeter at 22 °C. IR spectra were recorded on a Bruker Tensor 27 spectrophotometer. 1D and 2D NMR spectra were recorded either on a Bruker Avance 300 MHz, Bruker Avance III (1H at 400 MHz; 13C at 100 MHz) or a Varian Unity Inova 500 (1H at 500 MHz; 13C at 125 MHz) using CDCl3 as solvent and TMS as internal standard. EIMS were recorded on a JEOL JMS-SX102 A mass spectrometer. FABMS spectra were determined on a JEOL Mstation JMS-700 mass spectrometer, using dinitrobenzyl alcohol as matrix. DARTMS and HRDARTMS were measured on a JEOL AccuTOF JMS-T100LC. Analytical thin layer chromatography (TLC) was performed on precoated Alugram® Sil G/UV254 plates, using UV and Ce(SO4)2/H2SO4 for spot detection. Preparative TLC was carried on Macherey-Nagel Sil G-200 UV UV254 plates (2.0 mm thickness). Vacuum-assisted column chromatography (VCC) was performed on silica gel 60 G (Macherey-Nagel), unless another stationary phase, such as Tonsil Clay Acticil 220-FF (Química Rique S. A.), is specified.
Plant material
Aerial parts of Physalis glutinosa Schltdl. were collected in autumn, in October 2014, in San Felipe, Guanajuato State (N21°28'14”, W101°15'40” at 2390 masl), and those of Physalis latiphysa Waterf. in summer, in August of 2016, between San Miguel de Allende and Dolores Hidalgo, Guanajuato State (N21°07'20”, W100°49'52” at 2390 masl), Mexico. The plants were identified by Dr. M. Martínez, and voucher specimens were deposited in the Herbarium of the Universidad Autónoma de Querétaro, P. glutinosa (QMEX 5442), P. latiphysa (QMEX 14988).
Extraction and isolation
P. glutinosa
Dried and milled leaves, flowers, and stems of the plant (542.4 g) were successively extracted with hexane, acetone and MeOH. After solvent evaporation, 65.4, 10.3, and 44.1 g of extracts, respectively, were obtained. The hexane extract was fractioned by VCC eluted with hexane-EtOAc mixtures of increasing polarity (1:0 to 2:3 v/v) to obtain fractions A1-A7(1:0); A8-A39 (19:1); A40-A80 (9:1); A81-A100 (17:3); A101-A106 (4:1); A107-A114 (7:3); A115-A124 (6:4); A125-A131 (2:3) and A132 (Me2CO). Fractions A2-A24 (25.52 g) were purified by VCC using a gradient of hexane-EtOAc (1:0 to 7:3 v/v) to afford fractions B1-B5 (1:0); B6-B20 (39:1); B21-B31 (19:1); B32-B37 (9:1); B38-B52 (17:3); B53-B63 (4:1) and B64-B71 (7:3). Crystallization (EtOAc-hexane) of fractions B30-B38 gave 83.7 mg of retusin (8). The mother liquors of 8 (1.65 g) were purified by VCC eluted with CHCl3-EtOAc to obtain fractions C1-C4 (1:0) and C5-C17 (49:1). Fractions C10-C17 gave 28.3 mg of 12-O-acetylphysacoztomatin (3). Fractions C1-C7 (662 mg) were subjected to VCC eluted with a gradient of hexane-CHCl3-Me2CO to afford fractions D1-D18 (1:4:0); D19-D24 (1:9:0); D25-D27 (0:1:0); D28-D41 (0:49:1); D42 (0:0:1). Crystallization (EtOAc-hexane) of fractions D8-D11 and A25-A39 gave 52.9 and 77.9 mg of compound 8, respectively. The mother liquors of 8 and fractions A40-A56 were combined (14.26 g) and subjected to VCC eluted with hexane-Me2CO to obtain fractions E1-E36 (49:1); E37-E41 (19:1); E42-E46 (9:1); E47-E49 (17:3). Fractions E7-E12 gave 47.5 mg of compound 8 for a total yield of 262 mg (0.048 % based on the weight of dry plant material). VCC of fractions E14-E49 (8.54 g) using a gradient of hexane-Me2CO (49:1 to 17:3) gave fractions F1-F27. Fractions F8-F25 (eluted with hexane-Me2CO 9:1 to 3:1) contained physaglutinic acid (1). Crystallization of fractions A57-A85 (EtOAc-hexane) afforded 3.82 g of 12-epi-nicandrodiol (6). The mother liquors of compound 6 were pooled with fractions A86-A120, B39-49 and D12-D30 (12.94 g) and subjected to VCC using as eluent hexane-Me2CO mixtures of increasing polarity to afford fractions G1-G43 (4:1); G44-G49 (3:1); G50-G52 (7:3). Fractions G8-G12 gave 4.01 g of compound 6. Fractions G4-G7 were fractioned on a Sephadex LH-20 column eluted with MeOH to obtain fractions H1-H16. Fraction H-4 (1.635 g) was subjected to VCC eluted with hexane-Me2CO 4:1 to obtain fractions I1-I95 from which, fractions I79–I95 were subjected to VCC (eluent hexane-Me2CO 4:1) to afford fractions J1-J16, then, fractions J2-J4 (202.6 mg) were purified by VCC using hexane-iPrOH 99:1 as eluent to give fractions K1-K25. Preparative TLC (hexane-Me2CO 7:3) of fractions K3-K9 (89.0 mg) afforded 28.4 mg of 1. Fractions A121-A131 and G13-G52 were combined (1.88 g) and subjected to VCC eluted with CHCl3-EtOH 99:1 to give fractions L1-L34. VCC (eluent hexane-Me2CO 33:7) of fractions L1-L23 (431.5 mg) gave fractions M1-M13. All fractions containing compound 1 were mixed (F8-F27, I64-I78, K10-K17, and M7-M13) and subjected to VCC eluted with hexane-Me2CO 4:1 to afford fractions N1-N134. Fractions N43-N61 dissolved in EtOAc were extracted with 5% Na2CO3; the aqueous phase was acidified with 5% HCl and extracted with EtOAc. This organic phase was washed with H2O and dried with anhydre Na2SO4 to afford, after solvent evaporation, 4.76 g of a residue, which was purified by two consecutive VCC (eluents hexane-Me2CO 9:1 and hexane-iPrOH 98:2) to obtain 1.633 g of semipure compound 1, a portion of which (197.6 mg) was finally purified by VCC (Tonsil Clay Acticil 220-FF; hexane-iPrOH 97:3 as eluent) to afford 154.8 mg of pure physaglutinic acid (1). The acetone extract was fractioned by VCC using a gradient of hexane-EtOAc (1:0 to 0:1 v/v) to obtain fractions Q1-Q102, from which, fractions Q36-Q46 (eluted with hexane-EtOAc 4:1 to 3:2; 2.78 g) were purified by VCC using mixtures of hexane-EtOAc (4:1 to 1:1) as eluents, to afford fractions R1-R98. Crystallization (EtOAc-hexane) of fractions R25-R43 (eluted with hexane-EtOAc 3:1 and 7:3) gave 266 mg of compound 6. In a similar way, the MeOH extract (44.14 g) was fractioned by VCC eluted with mixtures of hexane-EtOAc-MeOH (1:0:0 to 0:1:1) to give fractions S1-S98. Fractions S5-S13 (eluted with hexane-EtOAc-MeOH 4:1:0; 53.0 mg) were purified by VCC eluted with a gradient of hexane-EtOAc to afford fractions T1-T6 (4:1); T7-T10 (3:1), and T11-T14 (7:3). Fractions T4-T5 were crystallized from EtOAc-hexane to give 9.8 mg of physacoztomatin (4).
P. latiphysa
Fresh leaves, flowers, and stems of the plant (2.82 kg) cut in small pieces were macerated overnight with acetone ( ∼ 5 L, twice), then with MeOH ( ∼ 5 L, twice), and finally with a mixture of hexane-EtOAc 1:1 ( ∼ 3 L, twice). After solvents evaporation, the extracts were mixed and partitioned with EtOAc-H2O to afford 59.0 and 138.0 g of EtOAc and H2O extracts, respectively. During the partition process, rutin (17) precipitated from the aqueous phase as an amorphous yellow solid, which was purified by crystallization from MeOH (yield 7.98 g). The EtOAc extract was fractioned by VCC eluted with hexane-EtOAc mixtures of increasing polarity (1:0 to 3:2 v/v) to obtain fractions A1-A14 (1:0); A15-A24 (19:1); A25-A78 (9:1); A79-A101 (4:1); A102-A111 (7:3); A112-A125 (3:2); A126 (Me2CO). Fractions A18-A57 (11.7 g) were subjected to VCC using a gradient of hexane-EtOAc (19:1 to 11:9 v/v) to afford fractions B1-B50 (19:1); B51-B67 (37:3); B68-B94 (9:1); B95-B102 (17:3); B103-B109 (3:1); B110-B114 (13:7); B115-116 (11:9). Crystallization of fractions B19-B31 (EtOH) gave 212.5 mg of β-sitosterol. Fractions B54-B81 (2.55 g) were subjected to VCC eluted with mixtures of hexane-EtOAc to afford fractions C1-C29 (17:3) and C30-C37 (4:1). Fractions A58-A78 (2.653 g) were fractioned by VCC eluted with hexane-EtOAc mixtures to obtain fractions D1-D20 (4:1); D21-D28 (3:1); D29-D35 (7:3). Crystallization of fractions D13-D35 (EtOAc-hexane) gave 126.7 mg of labdenediol (11). Fractions B110-115, C7-C17 and mother liquors of fractions D-20-D24 were mixed (5.77 g) and purified by VCC using mixtures of hexane-EtOAc as eluents to obtain fractions E1-E52 (17:3); E53-E60 (4:1); E61-E80 (7:3). Fractions E61-E80 were discolored with activated charcoal and a portion (153.3 mg) were purified by preparative TLC (hexane-iPrOH 19:1, thrice) to obtain 30.7 mg of the physanicantriols (12/13). Crystallization of fractions A79-A96 (EtOAc-hexane) afforded 2.11 g of (+)-labda-8(17),13(Z)-dien-15,16-diol (9). The combined mother liquors of compounds 9 and 11 (2.94 g) were discolored with activated charcoal and subjected to VCC eluted with mixtures of hexane-acetone to afford fractions F1-F29 (9:1); F30-F45 (17:3); and F46-F70 (4:1). Fractions F31-F43 were purified by two successive VCCs using as eluent a mixture of hexane-iPrOH 97:3 and crystallization to give 739.0 mg of compound 9 and 287.4 mg of 11, for total yields of 2.849 g and 414.1 mg, respectively. Fractions A112-A126 were purified by VCC using as eluent mixtures of hexane-EtOAc-MeOH of increasing polarity to obtain fractions G1-G44 (1:1:0); G45-G54 (2:3:0); G55-G69 (1:4:0); G70-G76 (0:1:0); G77-G87 (0:19:1); G88-G95 (0:9:1). Fractions G4-G12 (1.45 g) were discolored with activated charcoal and purified by VCC using mixtures of hexane-acetone 7:3 as eluent to obtain fractions H1-H27. VCC (eluent hexane-acetone 4:1) of fractions H5-H11 (151 mg) gave fractions I1-I18, from which, fractions I5-I11 afforded 94.2 mg of nicandrose B (15). Fractions H14-H27 (210 mg) were purified by VCC eluted with hexane-iPrOH 9:1 to give fractions J1-J22. Fractions J14-J19 gave 57.1 mg of nicandrose D (16).
Physaglutinic acid (1)
Colorless gum; [α]20D + 5 (c 0.20, CHCl3); IR (CHCl3) νmax 3600-2400, 1731, 1660 sh, 1603, 1448, 1372, 1136, 1056, 1028 cm−1; 1H and 13C NMR: see Table 1; HRDARTMS m/z 466.31595: [M + NH4]+, C26H40O6NH4 (calcd. 466.31686).
Methylation of physaglutinic acid (1)
A solution of semi-pure compound 1 (116.5 mg) in MeOH (10 ml) was treated with CH2N2. The reaction mixture was stirred by 15 min. and the solvent was evaporated to yield 109.8 mg of the reaction mixture, which was purified by VCC eluted with hexane-Me2CO 9:1 to obtain 72.4 mg of methyl physaglutinate (2): Colorless gum; [α]20D + 11 (c 0.18, CHCl3); IR (CHCl3) νmax 1733, 1602, 1440, 1370, 1163, 1024, 994 cm−1; 1H and 13C NMR: see Table 1; FABMS m/z: 463 [M + H]+, 403 [M + H-AcOH]+, 331 [M + H - C5H8O4]+, 271 [M + H - C5H8O4 - AcOH]+.
Oxidation of compound 6
Pyridinium chlorochromate (261.8 mg) was added to a solution of 6 (100.7 mg) in CH2Cl2 (10 mL). The suspension was stirred at rt by 24 h, later it was purified by VCC eluted with hexane-CH2Cl2 4:1 and crystallization to afford 6.3 mg of pumiloxide (7): Colorless crystals; mp 87–88 °C; [α]20D -19 (c 0.22, CHCl3); [lit [5] mp 86–90 °C; [α]20D -21 (c 0.2, CHCl3)]. IR (CHCl3) νmax: 1643, 1511, 1460, 1387, 1163, 1150, 1090, 968, 890 cm−1; DARTMS m/z: 591 [2 M + NH4]+, 573 [2 M + H]+, 287 [M + H]+, 269 [M + H - H2O]+.
Acetylation of compounds 9
Compound 9 (44.8 mg) was treated with pyridine (0.5 mL) and acetic anhydride (0.5 mL). The reaction mixture was stirred at rt by 30 min and worked out in the usual way to obtain crude diacetyl derivative 10 (55.0 mg), which was purified by VCC (eluent hexane-EtOAc 19:1) to give compound 10 (50.6 mg): colorless oil; [α]20D + 2 (c 0.19, CHCl3); IR (film) νmax: 1743, 1672, 1643, 1459, 1443, 1368, 1226, 1026, 966, 889 cm−1; DARTMS m/z: 408 [M + NH4]+(12), 391 [M + H]+ (32), 331 [M + H - AcOH]+ (54), 271 [M + H - 2AcOH]+ (100); 1H and 13C NMR: see Table 2.
Oxidation of compound 9
Pyridinium chlorochromate (99.6 mg) was added to a solution of 9 (42.5 mg) in CH2Cl2 (10 mL). The suspension was stirred at rt by 30 min, then filtered through a Si gel G column with CH2Cl2 as eluent to afford 34.0 mg of a residue which was purified by a VCC eluted with hexane to obtain 27.1 mg of compound 14: Colorless oil [α]20D + 41 (c 0.34, CHCl3) [lit [23] [α]20D + 44.1 (c 0.59, CHCl3)]; IR (CHCl3) νmax 1641, 1602, 1521, 1475, 1440, 1424, 1024, 928, 893, 874 cm−1; DARTMS m/z: 573 [2 M + H]+ (6), 287 [M + H]+ (100).
Lipase inhibition assay
A previously reported colorimetric assay based on the hydrolysis of p-nitrophenylpalmitate was conducted with modifications [24]. Compounds were dissolved in a mixture of acetonitrile-ethanol (33.3:66.7) to achieve a 5 mM concentration and were kept in refrigeration (4 °C) upon use. Then, the following solutions were put in a 96-well microplate (added in order): 75 mM Tris-HCl buffer (pH 8.5), in a volume ranging from 50 to 80 μL, 10 μL of p-nitrophenylpalmitate (3 mM), test compound in a volume ranging from 1 to 30 μL, and 10 μL of type II porcine pancreatic lipase (EC 3.1.1.3; prepared at 10 mg/mL in 75 mM Tris-HCl buffer, pH 8.5). Final volume of each well was 100 μL. This mixture was incubated at room temperature for 5 min and afterwards, at 37 °C for 25 min. Then, absorbance was determined at 405 nm using a Synergy HT Multi-Mode Microplate Reader (Biotek, Winooski, VA, USA). Negative control consisted of 80 μL of buffer, 10 μL of substrate, and 10 μL of enzyme. Orlistat was used as a positive control (200 μM in ethanol). Final concentrations of evaluated compounds ranged from 0.3 to 300 μM, and then from 50 to 500 μM for compound 16. Six independent curves were constructed to obtain six replicates of each concentration. Orlistat concentrations used ranged from 0 to 200 μM. Percentage of inhibition was calculated using the following equation:
Where Anegative control is the mean absorbance of the wells used as negative control and A sample is the absorbance of each well. With the percentage of inhibition, the IC50 values were calculated using a non-linear regression using GraphPad Prism 9.0 software (GraphPad Inc., US).
Molecular docking
Crystal structure of the pancreatic lipase in complex with colipase and inhibited by an alkyl phosphonate (methoxyundecylphosphinic acid) was retrieved from the Protein Data Bank under the PDB ID 1LPB (https://www.rcsb.org/structure/1LPB). Chain B was used for docking simulation since it holds the catalytic site. The chemical models of the ligands (4, 10 and 16) were built with MOE (Molecular Operating Environment, v. 2022.02) and then subjected to energy minimization with a single point procedure using the MMFF94x force field implemented in the same software. In addition, the positive control orlistat and the co-crystallized inhibitor methoxyundecylphosphinic acid were also docked as reference. The docking involves a two-step protocol; first a placement algorithm where the ligands are placed in the site of interaction (conformation and fit are evaluated) and second, a refinement method to rank the final poses. The placement method was carried out using the triangle matcher algorithm, and the refinement was achieved using an induced fit method.
References
Zhang WN, Tong WY. Chemical constituents and biological activities of plants from the genus Physalis. Chem Biodivers. 2016;13:48–65. https://doi.org/10.1002/cbdv.201400435
Zhang H, Samadi AK, Cohen MS, Timmermann BN, Zhang W-N, Ong W-Y, et al. Antiproliferative withanolides from the Solanaceae: A structure-activity study. Pure Appl Chem. 2012;84:1353–67. https://doi.org/10.1021/np060274l
Pérez-Castorena AL, Oropeza RF, Vázquez AR, Martínez M, Maldonado E. Labdanes and withanolides from Physalis coztomatl. J Nat Prod. 2006;69:1029–33. https://doi.org/10.1021/np0601354
Pérez-Castorena AL, Martínez M, Maldonado E. Labdanes and sucrose esters from Physalis sordida. J Nat Prod. 2010;73:1271–6. https://doi.org/10.1021/np100127k
Torres FR, Pérez-Castorena AL, Arredondo L, Toscano RA, Nieto-Camacho A, Martínez M, et al. Labdanes, Withanolides, and Other Constituents from Physalis nicandroides. J Nat Prod. 2019;82:2489–500. https://doi.org/10.1021/acs.jnatprod.9b00233
Maldonado E, Pérez-Castorena AL, Romero Y, Martínez M. Absolute configuration of labdane diterpenoids from Physalis nicandroides. J Nat Prod. 2015;78:202–7. https://doi.org/10.1021/np500688c
Clifford DJ, Hatcher PG. Structural transformations of polylabdanoid resinites during maturation. Org Geochem. 1995;23:407–18. https://doi.org/10.1016/0146-6380(95)00022-7
Dentali SJ, Hoffmann JJ, Jolad SD, Timmermann BN. Diterpenes of Ericameria linearifolia. Phytochemistry. 1987;26:3025–8. https://doi.org/10.1016/S0031-9422(00)84585-9
Anderson KB, Winans RE, Botto RE. The nature and fate of natural resins in the geosphere—II. Identification, classification and nomenclature of resinites. Org Geochem. 1992;18:829–41. https://doi.org/10.1016/0146-6380(92)90051-X
Poulin J, Helwig K. Inside amber: The structural role of succinic acid in class Ia and class Id resinite. Anal Chem. 2014;86:7428–35. https://doi.org/10.1021/ac501073k
World Health Organization. World Obesity Day 2022 – Accelerating action to stop obesity. 2022. https://www.who.int/news/item/04-03-2022-world-obesity-day-2022-accelerating-action-to-stop-obesity Accessed 15 May 2023.
Kumar A, Chauhan S. Pancreatic lipase inhibitors: The road voyaged and successes. Life Sci. Pergamon; 2021. 119115. https://doi.org/10.1016/j.lfs.2021.119115.
Ferrulli A, Terruzzi I, Senesi P, Succi M, Cannavaro D, Luzi L. Turning the clock forward: New pharmacological and non pharmacological targets for the treatment of obesity. Nutr Metab Cardiovasc Dis. 2022;32:1320–34. https://doi.org/10.1016/j.numecd.2022.02.016
Jalaja R, Leela SG, Valmiki PK, Salfeena CTF, Ashitha KT, Krishna Rao VRD, et al. Discovery of natural product derived labdane appended triazoles as potent pancreatic lipase inhibitors. ACS Med Chem Lett. 2018;9:662–6. https://doi.org/10.1021/acsmedchemlett.8b00109
Yoshioka Y, Yoshimura N, Matsumura S, Wada H, Hoshino M, Makino S, et al. α-Glucosidase and pancreatic lipase inhibitory activities of diterpenes from indian mango ginger (Curcuma amada roxb.) and its derivatives. Molecules. 2019;24:4071. https://doi.org/10.3390/molecules24224071
Silva TMS, Carvalho MG, Braz-Filho R. Estudo espectroscópico em elucidação estrutural de flavonoides de Solanum jabrense Agra & Nee e S. paludosum Moric. Quim Nova. 2009;32:1119–28. https://doi.org/10.1590/S0100-40422009000500008
Villamizar J, Fuentes J, Salazar F, Tropper E, Alonso R. Facile access to optically active labdane-type diterpenes from (+)-manool. Synthesis of (+)-coronarin E, (+)-15,16-epoxy-8(17),13(16),14-labdatriene, and (+)-labda-8(17),13(Z)-diene-15,16-diol. J Nat Prod. 2003;66:1623–7. https://doi.org/10.1021/np030166o
Calabuig MT, Cortés M, Francisco CG, Hernández R, Suárez E. Labdane diterpenes from Cistus symphytifolius. Phytochemistry. 1981;20:2255–8. https://doi.org/10.1016/0031-9422(81)80124-0
Forster PG, Ghisalberti EL, Jefferies PR. Labdane diterpenes from an Acacia species. Phytochemistry. 1985;24:2991–3. https://doi.org/10.1016/0031-9422(85)80042-X
Maldonado E, Torres FR, Martínez M, Pérez-Castorena AL. Sucrose esters from the fruits of Physalis nicandroides var. attenuata. J Nat Prod. 2006;69:1511–3. https://doi.org/10.1021/np060274l
Kazuma K, Noda N, Suzuki M. Malonylated flavonol glycosides from the petals of Clitoria ternatea. Phytochemistry. 2003;62:229–37. https://doi.org/10.1016/S0031-9422(02)00486-7
Cambie RC, Moratti SC, Rutledge PS, Weston RJ, Woodgate PD. A Synthesis of (-)-12,15-epoxylabda-8(17),12,14-trien-16-yl acetate and (-)-pumiloxide. Aust J Chem. 1990;43:1151–62. https://doi.org/10.1071/CH9901151
Miyake T, Uda K, Kinoshita M, Fujii M, Akita H. Concise syntheses of coronarin A, coronarin E, austrochaparol and pacovatinin A. Chem Pharm Bull. 2008;56:398–403. https://doi.org/10.1248/cpb.56.398
García-De La Cruz L, Galvan-Goiz Y, Caballero-Caballero S, Zamudio S, Alfaro A, Navarrete A. Hypericum silenoides Juss. and Hypericum philonotis Cham. & Schlecht. extracts: In-vivo hypolipidaemic and weight-reducing effects in obese rats. J Pharm Pharmacol. 2013;65:591–603. https://doi.org/10.1111/jphp.12015
Birari RB, Bhutani KK. Pancreatic lipase inhibitors from natural sources: unexplored potential. Drug Discov. Today. Elsevier Current Trends; 2007. 879–89. https://doi.org/10.1016/j.drudis.2007.07.024.
Egloff MP, Marguet F, Buono G, Verger R, Cambillau C, van Tilbeurgh H. The 2.46Å resolution structure of the pancreatic lipase-colipase complex inhibited by a Cll alkyl phosphonate. Biochemistry 1995;34:2751–62. https://doi.org/10.1021/bi00009a003
Bernal CA, Castellanos L, Aragón DM, Martínez-Matamoros D, Jiménez C, Baena Y, et al. Peruvioses A to F, sucrose esters from the exudate of Physalis peruviana fruit as α-amylase inhibitors. Carbohydr Res. 2018;461:4–10. https://doi.org/10.1016/j.carres.2018.03.003
Mora Vargas JA, Orduña Ortega J, Metzquera G, Larrahondo JE, Boscolo M. Natural sucrose esters: perspectives on the chemical and physiological use of an under investigated chemical class of compounds. Phytochemistry 2020;177:112433. https://doi.org/10.1016/j.phytochem.2020.112433
Acknowledgements
We are grateful to R. Gaviño, I. Chávez, A. Peña, and E. Huerta for the NMR spectra; R. Patiño for the IR and optical rotations, and C. García, J. Pérez, and C. Márquez, for the MS. B. Ovalle thanks Dirección General de Asuntos de Personal Académico, UNAM for the grant (DGAPA-UNAM IA205621).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maldonado, E., Mineros, A., Torres, O.D. et al. Lipase inhibitory activity of constituents of Physalis glutinosa and Physalis latiphysa. Med Chem Res 32, 2505–2515 (2023). https://doi.org/10.1007/s00044-023-03151-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03151-6